Article Text

Statistics from Altmetric.com
Introduction
Neuromyelitis optica (NMO) is a relapsing demyelinating disorder with a predilection for the optic nerves and spinal cord, associated with autoantibodies to aquaporin-4 (AQP4) water channels in most cases. It affects all races and ages, but has a striking female preponderance and is distinct from multiple sclerosis (MS).
This is a pragmatic guide to treating NMO and the associated NMO spectrum disorders (NMOSDs); these include limited forms of the syndrome, such as recurrent longitudinally extensive transverse myelitis (LETM), recurrent severe optic neuritis (ON) or atypical presentations associated with serum AQP4 antibodies. Because these disorders are rare, there is no available evidence from randomised controlled trials. In contrast to MS, NMO has a high early morbidity and mortality because of severely disabling relapses and lacks a progressive phase. For example, studies report death in 25–30% of patients, after a mean of 5 years from onset.1 ,2 About half of the patients develop significant walking difficulties (at a mean time from onset of 7 years) and many patients become dependent on wheelchairs (figure 1). Visual impairment is also common, with blindness affecting at least one eye in 60–70%, at a mean time from onset of 5 years.1–3 Although most available studies (observational and retrospective) include patients treated at various times from onset, immunosuppressive treatments clearly dramatically reduce relapses, that is, around a sixfold reduction compared with pretreatment.4–8 Thus, aggressive treatment of relapses and effective prevention of relapses is the prime aim of treatment and is our single most important message.
For the purposes of this review, we assume that the diagnosis of NMO or NMOSD is correct, and that similar disorders such as MS (for which the treatment is different) are excluded.
The rarity of NMO and the importance of optimising treatment makes specialist input important. In the UK, there is a nationally commissioned NMO service, providing a free multidisciplinary clinical and telephone advisory service. The direct telephone helplines (+44 1515298357 and +44 1865231905) and UK NMO website www.nmouk.nhs.uk can help patients, their families, doctors and other healthcare workers with further information.
Principles of management
As they are both treated in the same way, we refer to NMO and NMOSD under the umbrella term of NMOSDs. We refer to the first event as the onset attack and subsequent attacks as relapses. The treatment paradigms are similar to other antibody-mediated conditions, such as myasthenia gravis (MG). Our treatment regimens apply to adults, although similar principles apply to children, with input from paediatric physicians.
From the onset it is crucial to establish that the patient understands the aims of treatment: prevention of relapses and effective treatment of relapses to limit disability resulting from any relapses that do occur. This requires patients’ urgent reporting of relapses and their concordance with immunosuppressive therapy. It is important to be open about the irreversibility of longstanding neurological deficits and the role of symptomatic treatments.
In contrast to the European guidelines9 we (a) do not recommended rituximab as first line and (b) recommend immunosuppressive therapy in limited phenotypes with AQP4 antibodies, whatever the relapse frequency or severity, because of the high risk of relapses and disability in the future.10 ,11 In fact, approximately one-third of our patients with relapsing NMO have a non-severe onset attack.
There are three aspects to treatment:
-
Relapse treatment
-
Relapse prevention
-
Symptom management and rehabilitation.
Relapse treatment
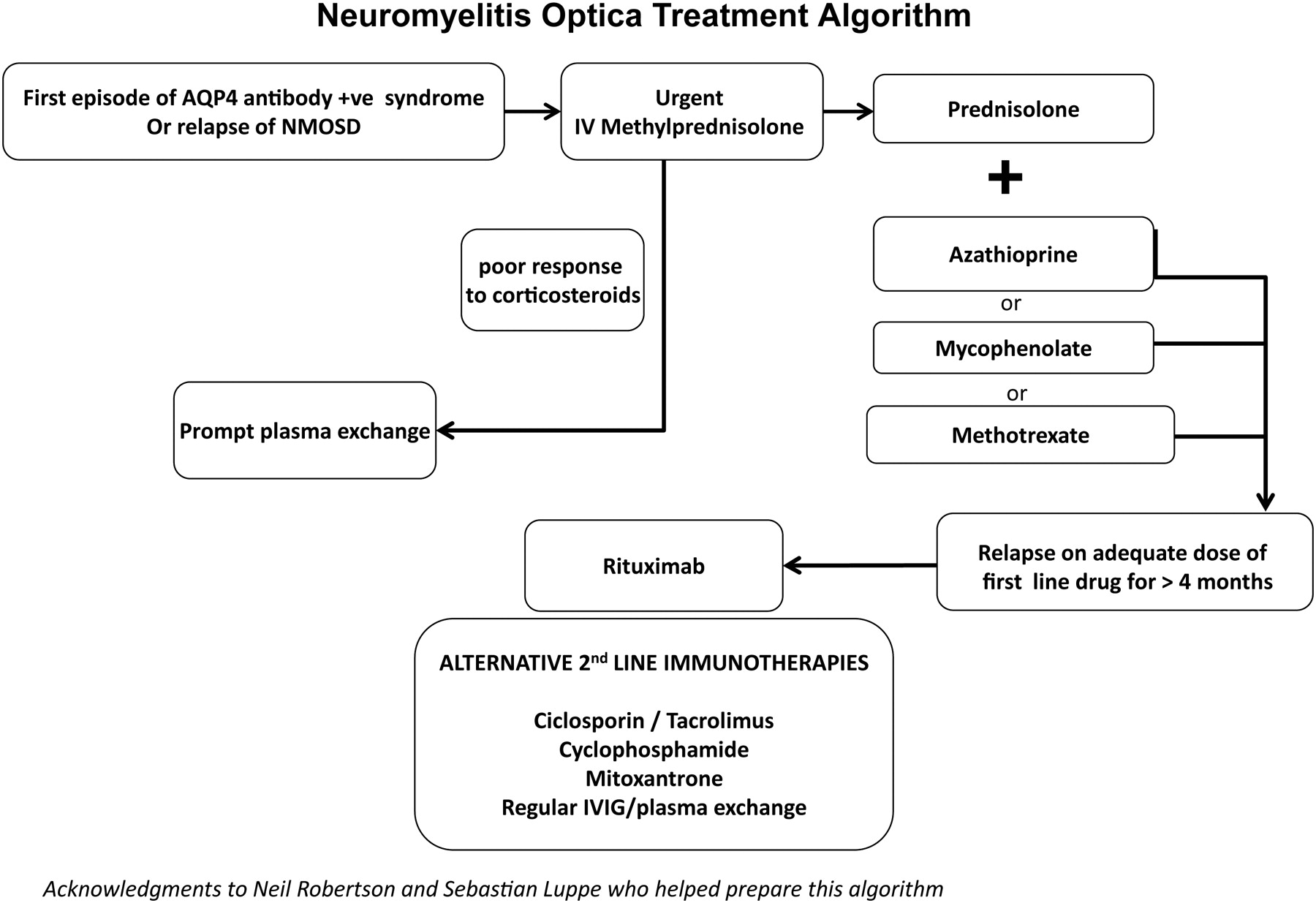
Patients with NMOSD relapses tend to respond to intravenous corticosteroids; because relapses can lead to severe and permanent disability, we recommend prompt treatment. This requires a fast-track access plan for the patient, which must be communicated to the family, general practitioner and hospital teams. Intravenous methylprednisolone (1 g daily for 3–5 days) is the conventional treatment and in most patients this will give some recovery. Corticosteroid poorly responsive relapses should be treated with plasma exchange, started quickly. We recommend plasma exchange if there is no response within 5 days of starting methylprednisolone or 7–10 days if there is a partial but inadequate response. However, marked improvements with plasma exchange can occur even several weeks later.
Patients should remain on oral prednisolone as relapse (or rebound) prevention, after their acute treatment. Even in those who do not require long-term immunosuppression (eg, monophasic antibody-negative patients), early rebound may follow if corticosteroids are stopped quickly. This is distinct from a relapse (the same situation as in acute disseminated encephalomyelitis). We therefore recommend an oral weaning course of prednisolone over 2–6 months. Myelitis-related disability often improves over the subsequent 2–3 years, if the patient remains relapse free.
Relapse prevention
Patients requiring relapse prevention (Box 1) should receive immunosuppressive (or immunomodulatory) treatment. This should follow the onset attack if they are AQP4 antibody-positive or if they fulfil the NMO criteria. Patients who have had only a single attack of LETM or ON (bilateral or severe) but who are antibody-negative need treatment only if they relapse; their lower risk of relapse allows them the chance to avoid the risks of long-term immunosuppression. The general principle is to achieve remission quickly with corticosteroids and then slowly to reduce the corticosteroid dose once the steroid-sparing immunosuppressive therapy becomes effective, thereby reducing the risks of long-term side effects.
Box 1 Who should be given immunosuppressive treatment?
Patients
-
With aquaporin-4 (AQP4) antibodies*
-
Without antibodies but fulfil the diagnostic criteria for neuromyelitis optica (NMO)*
-
optic neuritis and longitudinally extensive transverse myelitis
-
brain MRI not diagnostic for multiple sclerosis (MS)
-
-
Optico-spinal demyelination atypical for MS or NMO
-
Relapsing severe optic neuritis or myelitis (longitudinally extensive transverse myelitis) without alternate diagnosis and not fulfilling MS criteria
*Antibodies should be measured in an experienced laboratory.
Corticosteroid treatment
Daily or alternate-day regimens (at equivalent doses) of prednisolone are commonly used at a starting dose of up to 1 mg/kg: usually around 50–60 mg daily. This should be reduced to a maintenance dose over 6 months while first-line steroid-sparing agents are taking effect. After this period, if the patient remains stable, it is reasonable to taper the dose of prednisolone further. The safe lower limit of maintenance corticosteroid therapy can only be found by gradual taper, with the attendant risk precipitating a relapse. Although this is acceptable in some other antibody-mediated conditions such as MG, it is risky in NMOSD, where relapses can cause severe irreversible disability. Our impression is that NMO is particularly corticosteroid dependent. Some patients can wean off corticosteroids but it is not possible to predict this subgroup; we tend to continue long-term low-dose corticosteroids, if well tolerated, at around 10–20 mg daily (or an equivalent alternate-day regimen).
Antibody titre monitoring may prove to be useful for monitoring individual patients, particularly if tested at times relevant to treatment changes and during relapse, to compare with levels during stable periods (figure 2). Note, however, that across-patient comparisons of antibody titres do not predict disease severity or individual patient thresholds for relapse. Unlike MS, MRI has no role in monitoring treatment efficacy at present because subclinical activity seems uncommon. However, we do sometimes use gadolinium-enhanced MRI, combined with AQP4 antibody titres, to assess disease activity when trying to distinguish between relapses and pseudo-relapses (Box 2).
Box 2 Guidelines for immunosuppression with prednisolone and azathioprine for neuromyelitis optica spectrum disorders
Immunosuppressive therapy should be started at diagnosis, that is, usually during a relapse or onset attack. Thus, it will usually be introduced concurrently with acute treatment as an in-patient.
Prednisolone
-
Start 0.75–1.0 mg/kg daily for 1 month
-
Reduce by 5 mg every month
-
Maintain on 20 mg daily (or 40 mg alternate days)
-
Further reductions can be made if remain stable over the next 6 months, although many patients remain corticosteroid-dependent and, if well tolerated, low maintenance doses of 10–20 mg daily (or equivalent) can be continued
Azathioprine
-
Ensure thiopurine methyltransferase level is normal
-
Target dose = 2.5–3.0 mg/kg body weight per day, in two divided doses
-
Inpatient (IP): Start 25 mg and increase by 25 mg daily
-
Outpatient (OP): Start 25 mg and increase by 50 mg weekly
-
Check full blood count and liver function twice weekly as an IP, or weekly as OP, until on target dose for 1 month, then monthly (3-monthly when stable over the long term)
-
A red cell macrocytosis and/or lymphopenia is a useful marker of adequate dose

Relapses may leave patients wheelchair bound and visually impaired.
Steroid-sparing agents
We recommend starting add-on immunosuppression as steroid-sparing agents at the same time as corticosteroids.
Azathioprine is our first-line steroid-sparing agent. The dosing regimen is simple because there is a single initial target maintenance does of 2.5 mg/kg (better tolerated when divided into two daily doses), which can be increased further to 3 mg/kg. An elevated mean corpuscular volume or lymphopenia can indicate that the treatment is at a therapeutic level and, if not present, may suggest the need to increase the dose. It is good practice to measure thiopurine methyltransferase before starting; a low level is a contraindication to azathioprine. However, adverse effects can still occur with normal levels and azathioprine intolerance in NMO patients seems relatively common in others’ (22%)4 and our own experience.
Methotrexate is an alternative, particularly in patients who do not tolerate azathioprine. Family doctors often have experience in using methotrexate, through treating more common conditions such as psoriasis and rheumatoid disease. It may be quicker acting than azathioprine and the dose required is variable. We aim for an initial maintenance dose of 15 mg once weekly (starting 7.5 mg in week 1 and increasing by 2.5 mg per week), with folate supplementation. If relapses occur, we increase the dose in 2.5 mg doses to a maximum of 25 mg weekly. However, it is not suitable for women of childbearing age and may lower the sperm count in men; we therefore recommend stopping it 3 months before trying to conceive.
Mycophenolate mofetil is an effective alternative and may be quicker acting than azathioprine. We generally start it at 500 mg daily in week 1, then increased to 500 mg twice daily in week 2, then 1 g morning and 500 mg evening in week 3 and 1 g twice daily thereafter. It is relatively contraindicated in pregnancy because of an increased risk of first trimester pregnancy loss and of congenital malformations; this is supported by its teratogenic effect in animals (Box 3).
Box 3 Other immunosuppressive therapy
Usual adult target doses
-
Methotrexate (orally) 15–25 mg once weekly*
-
Mycophenolate (orally) 1 g twice daily*
-
Rituximab 1000 mg intravenous at days 1 and 14, repeated 6-monthly
*Blood test monitoring as for azathioprine. Baseline tests and monitoring for all should be in accordance with local policy and guidelines.
Second-line treatments
Although the European Federation of Neurological Societies’ guidelines suggest that rituximab may be a first-line steroid-sparing option, its high cost and the lack of head-to-head comparative data preclude its use in many countries. As most first-line drugs seem to work well at the right doses when combined with low-dose corticosteroids, and because conventional immunosuppressants have well-established side-effect profiles (allowing a more accurate discussion around risk), we use rituximab for those who relapse despite adequate first-line treatment for a reasonable period (ie, 6 months). The usual dosing regimen is 1 g intravenous on days 1 and 14, repeated every 6 months or alternatively, when the CD19 count begins to rise. Anecdotally, some patients relapse shortly after rituximab, emphasising the need for reasonable doses of prednisolone to cover the first 6 months of rituximab treatment. Rituximab does not work for all patients and other second-line options for more resistant cases include cyclophosphamide, mitoxantrone, ciclosporin, tacrolimus, pulsed plasma exchange or pulsed intravenous immunoglobulin (figure 3).

Shows the aquaporin-4 (AQP4) antibody titre in an individual patient over time, and relates this to relapses (myelitis and/or optic neuritis) and the treatment regime.

{kind=link}
{kind=link}
{kind=link}
Summarises the management of relapse treatment and prevention.
Cautions and risks
Conventional immunosuppressants have well-described and frequent side effects; however, serious adverse events are rare, particularly when following monitoring guidelines. We are increasingly aware of unforeseen adverse effects with newer immunomodulatory treatments, such as atypical infections (including progressive multifocal leucoencephalopathy) particularly when other immunotherapies have been used; we are also seeing therapy-related leukaemia and cardiomyopathy with mitoxantrone.
Thus, it is crucial to provide patients and their families with adequate information and to make opportunities for their questions before and during treatment. We recommend that neurologists who supervise the treatment have experience in using these drugs, have sufficient resources to monitor the patients adequately and that they maintain a high level of vigilance for serious side effects. We also recommend using these treatments at proper doses and for a reasonable time, rather than ‘jumping’ across multiple treatments.
Patients and their physicians often consider further reducing or stopping maintenance immunosuppressive therapy when patients have been stable for a while. In our view, withdrawing treatment is risky, particularly in those with AQP4 antibodies, because we cannot predict whose disease may recur. Thus, at present, we do not recommend immunosuppressant drug withdrawal.
Special circumstances
Pregnancy planning
Azathioprine, ciclosporin and prednisolone appear relatively safe in pregnancy. Pregnancy issues are important to bear in mind when treating girls in paediatric services, even though pregnancy is often not on the radar. Thus, even when patients are not planning pregnancy, clinicians should discuss its future potential with patients, their family or partners.
Postpartum relapse risk
In patients with AQP4 antibodies, the relapse rate increases 3–4-fold during the 6-month postpartum.12 Therefore, patients and doctors should be aware of that risk and ensure maintenance of preventive immunosuppression.
Treatment of ‘overlap syndromes’
There is a subgroup of AQP-4 antibody-negative relapsing patients whose phenotype gives difficulty in distinguishing between MS and NMOSD. A second opinion is useful in this situation. We treat this group with drugs effective for both MS and NMOSDs, such as azathioprine or mitoxantrone. We avoid interferon-β,13 natulizumab14 and fingolimod,15 which increase the likelihood of NMO relapse. Also, we avoid alemtuzumab because it commonly precipitates autoantibody diseases.
Symptom management
We will not address general symptom management that the neurologist is already familiar with, although two symptoms deserve special mention.
Vomiting and hiccups
Refractory and otherwise unexplained vomiting and hiccups may suggest a brainstem relapse, although it is often attributed to other causes. In an NMOSD patient, these symptoms justify investigating with MR brain imaging and treating as a relapse.
Transverse myelitis-associated pain and tonic spasms
Neuropathic pain arising from transverse myelitis in NMOSDs appears to be more severe and disabling than in MS16 and often does not respond to conventional treatment such as tricyclic antidepressants, gabapentin or pregabalin. It typically starts as recovery begins from the acute attack and can continue for years, having a large impact on quality of life. Thus we recommend involving the local pain team early.
Tonic spasms from transverse myelitis attacks are also more common than in MS. A small dose of carbamazepine is often very effective, as in MS; the symptoms subside over weeks or months, probably reflecting resolution of inflammation or remyelination. Alternatives such as oxcarbazepine, lamotrigine, gabapentin or pregabalin can help.
Future
Combining treatments or starting with an ‘induction regimen’ followed by maintenance therapy may prove useful. Future potential treatments include humanised anti-CD20 monoclonals (eg, ofatumumab and ocrelizumab), modulation of Th17 lymphocytes, glutamate receptor and B-cell activating factors, and even AQP-4 binding protective antibodies to name a few. However, there is a crucial need for evidence-based data to identify the best way to preventing disability and to persuade commissioning bodies to fund new and potentially expensive treatments. The next logical step is a pragmatic international study integrated into clinical practice (see www.nmouk.nhs/uk/clinical-trials).
Practice points
-
There are no randomised controlled trials in neuromyelitis optica (NMO). Therefore, treatment guidelines are based on retrospective studies and consensus opinion.
-
Relapses are usually severe and often lead to permanent disability if not treated promptly.
-
Aquaporin-4 (AQP4) antibody seropositive disease is associated with high risk of relapse.
-
AQP4 antibody-negative relapsing disease without an alternative diagnosis is best treated like NMO.
-
The selection of preventative therapies in overlap syndromes requires caution because some immunotherapies used in multiple sclerosis may exacerbate NMO.
-
Most patients can be relapse-free if kept on long-term immunosuppressive medication.
-
Adverse effects of immunosuppressive or immunomodulatory treatments are inevitable, although usually tolerable.
-
Patient information and general practitioner (GP) involvement are integral.
-
Patients need a simple and fast-track access plan for the urgent management of relapses, which also needs to be communicated to the family, GP and hospital teams.
-
We need further evidence to identify the best treatment regimens for NMO patients.
Key points
-
The selection of preventative treatments in overlap syndromes requires caution, because some immunotherapies used in multiple sclerosis appear to exacerbate neuromyelitis optica.
-
Patients need a simple and fast-track access plan for the urgent management of acute relapse, which also needs to be communicated to the family, general practitioner and hospital teams.
Acknowledgements
We would like to acknowledge the funding of the UK NMO service by the National Commissioning team.
References
Footnotes
-
Contributors All authors have contributed to the writing of this paper.
-
Provenance and peer review Commissioned; externally peer reviewed. This paper was reviewed by Neil Scolding, Bristol, UK.
Linked Articles
- Editors' choice
Other content recommended for you
- Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: practical considerations
- Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders
- Antibodies to MOG and AQP4 in children with neuromyelitis optica and limited forms of the disease
- Neuromyelitis optica spectrum disorder (NMOSD) presenting as acute transverse myelitis with positive aquaporin 4 antibodies
- Overlapping CNS inflammatory diseases: differentiating features of NMO and MS
- Relapsing neuromyelitis optica in an adolescent girl
- A practical approach to the diagnosis of spinal cord lesions
- Paediatric neuromyelitis optica: clinical, MRI of the brain and prognostic features
- The borderland of neuromyelitis optica
- Neurodegeneration in multiple sclerosis and neuromyelitis optica