Article Text

Statistics from Altmetric.com
The clinical practice of neuromuscular disease is currently undergoing enormous change as a direct result of the wealth of recent molecular genetic discoveries. Indeed, the majority of gene discoveries in the area of neurological disease relate to neuromuscular disorders. The immediate impact of these discoveries is that a precise DNA based diagnosis is possible. This often gives patients accurate prognostic and genetic counselling information. It will also facilitate rational screening programmes for recognised complications such as cardiac or respiratory involvement. Unfortunately, at present many eligible patients do not benefit from or have access to such diagnostic precision, although this is changing.
The discovery of new genes and proteins has opened up unexplored avenues of research into therapies for neuromuscular patients. While therapeutic trials in genetic neuromuscular diseases remain in their infancy, it seems clear that a precise DNA based diagnosis will be essential. Eligibility for such trials and indeed for future proven therapies will be contingent upon DNA based diagnosis. For example, it is no longer acceptable to make “limb-girdle muscular dystrophy” based on simple histochemistry, a final diagnosis. Detailed immunocytochemistry and protein chemistry in combination with DNA analysis offer the patient the best chance of a precise diagnosis from which accurate prognostication, screening, and genetic counselling will follow.
In this review we describe some of the more common genetic nerve and muscle diseases encountered by adult neurologists. We have elected not to include the spinal muscular atrophies or the neuromuscular junction disorders (see web review for references on these diseases). Our aim is not to give a detailed description of each disorder, but to inform the reader about the latest genetic information and particularly about the availability of DNA based diagnosis. We will describe how DNA based diagnosis may accelerate and simplify the diagnostic process, thereby providing the most accurate information for patients with these diseases.
GENETIC PERIPHERAL NERVE DISEASE
While the peripheral nervous system can be involved in many widespread neurological or multisystem inherited disorders, this review will concentrate on those where a neuropathy is the sole or predominant problem (table 1).
Classification of the inherited neuropathies
Charcot-Marie-Tooth diseases
The Charcot-Marie-Tooth (CMT) diseases, also known as the hereditary motor and sensory neuropathies (HMSN), are a clinically and genetically heterogeneous group of inherited neuropathies1. CMT is a common disease affecting children and adults with an estimated prevalence of 1 in 2500. CMT is characterised clinically by distal muscle wasting and weakness, reduced reflexes, and impaired distal sensation, and neurophysiologically by a sensory motor neuropathy. The severity of the disease varies enormously, the variation depending to a large extent on the underlying genetic defect. It can be difficult sometimes to differentiate some forms of CMT from hereditary sensory and autonomic neuropathies (HSAN) and distal hereditary motor neuropathy (HMN). The sensory involvement is usually more severe in HSAN and as many of the genes are known for HSAN, the diagnosis can often be resolved by genetic testing. The differentiation between CMT and distal HMN can be more difficult clinically as some CMT patients do not have sensory symptoms or signs. Therefore the key to the diagnosis here is the neurophysiology as patients with CMT will always have sensory involvement and patients with distal HMN should never have sensory involvement.
The classification of CMT is in a state of constant flux, reflecting the rapid advances in the description of causative genes. Although the term HMSN is still used in the clinical literature, the genetic literature favours CMT and this nomenclature is now in more widespread use. As all the causative genes for CMT have not been described a combination of a clinical and an incomplete genetic classification is used at the moment (table 2). Most current classifications agree that the basic division of CMT should be done by electrophysiological criteria into type 1 (demyelinating) and type 2 (axonal)2 and that further subclassification should be according to inheritance pattern (autosomal dominant, recessive or X linked). Further subdivisions are usually done numerically or alphabetically but these are not universally agreed except for the more common forms (for example, CMT1A, CMT1B). Table 2 uses an alphabetical subdivision but other frequently used terms and/or the terms used by the original authors are given in brackets for clarity.
Classification of Charcot-Marie-Tooth disease
Demyelinating CMT
CMT1 (demyelinating CMT) is diagnosed when the median nerve motor conduction velocity is less than 38 m/s. There are autosomal dominant, autosomal recessive, and X linked forms of CMT1. Despite CMT1 being a demyelinating neuropathy, it is worth remembering that the disability in CMT1 patients regardless of the type is related to axonal loss.
Autosomal dominant CMT1
Autosomal dominant CMT1 can be further subdivided into CMT1A, B, C, and D. Most patients with autosomal dominant CMT1 present in the first or second decade of life with slowly progressive distal wasting and weakness and hyporeflexia. Distal sensory loss and foot deformity are also frequent findings on examination. Foot deformities such as pes cavus may be particularly useful in suggesting the diagnosis in patients without a family history. Neurophysiologically, as well as the expected reduction in motor conduction velocities, the sensory action potentials (SAPs) are reduced or absent as in all forms of CMT.
CMT1A is the most common form of autosomal dominant CMT1 and is usually caused by a 1.5 Mb duplication of chromosome 17 (17p11.2).3 This duplication accounts for 70% of all CMT1 cases.4 The causative gene for CMT1A in the duplication is the peripheral myelin protein 22 gene (PMP-22), mutations of which can rarely cause CMT1A.
CMT1B is a less common form of autosomal dominant CMT1 and is caused by point mutations in the human myelin protein zero (P0) gene on chromosome 1q22-q23. A recent interesting discovery is that mutations in the P0 gene can occasionally give rise to an axonal neuropathy (CMT2).
The locus for CMT1C, which is also autosomal dominant, has recently been described on chromosome 16p13.1-p12 but the causative gene has yet to be identified.
CMT1D is a rare form of CMT1 caused by mutations in the early growth response 2 gene (EGR2) on chromosome 10.
Hereditary neuropathy with liability to pressure palsies
A deletion of the same 1.5 Mb portion of chromosome 17, duplicated in CMT1A, is usually responsible for hereditary neuropathy with liability to pressure palsies (HNPP), an autosomal dominant condition that produces an episodic, recurrent, demyelinating neuropathy. Rarely, point mutations in PMP-22 cause HNPP. The typical clinical presentation of HNPP is of recurrent transient pressure palsies with a mild background demyelinating neuropathy neurophysiologically. HNPP can present with atypical features including a scapulo-peroneal syndrome and recurrent focal transient sensory symptoms, which may cause initial diagnostic difficulties, but usually patients have a generalised patchy demyelinating neuropathy neurophysiologically.
X linked CMT1
X linked CMT1 (CMT X1) is the X linked form of CMT1 and is caused by point mutations in the connexin 32 gene (Cx32). Affected male patients may present in an indistinguishable way from CMT 1A but are more likely to have median motor conduction velocities in the intermediate range (25–45 m/s). The nerve conduction studies in CMT X1 sometimes show a more patchy involvement than would be expected in CMT. There is also a higher incidence of mild central nervous system involvement (such as abnormal brainstem evoked potentials), which is often asymptomatic. “Carrier females” are often mildly affected and may have motor conduction velocities in the axonal range.
Dejerine Sottas disease and congenital hypomyelinating neuropathy
Dejerine Sottas disease (DSD) and congenital hypomyelinating neuropathy (CHN) are more severe demyelinating hypomyelinating neuropathies which usually present in the first decade with extremely slow motor conduction velocities and a more severe neuropathy than that seen with autosomal dominant CMT1. Although the terms DSD and CHN are still useful clinically, both diseases are now considered to be more severe forms of CMT1 and are most commonly caused by de novo point mutations of the three genes (PMP-22, P0, and EGR2) that normally cause CMT1. As you can see in table 2, in DSD A, DSD B, and CHN C, point mutations in the relevant genes, PMP-22, P0, and EGR2 can exist in the heterozygous or homozygous state, meaning DSD A, DSD B, and CHN C can be autosomal dominant or recessive.
Autosomal recessive CMT1
The identification of the causative genes in autosomal recessive CMT1 (AR-CMT1) is a rapidly expanding field, especially in the last few years, but as in any rapidly moving field the classification is in a state of flux (table 2). There are currently eight loci (excluding the three genes associated with autosomal recessive DSD and CHN) and four genes described.
AR-CMT1 A (CMT4A), an autosomal recessive form of CMT1, has recently been shown to be caused by mutations in the ganglioside induced, differentiation associated protein-1 (GDAP1). This is a particularly interesting gene as it has also been described to cause an autosomal recessive axonal form of CMT (see below).
AR-CMT1 B1 (CMT4B1) is an autosomal recessive demyelinating neuropathy with focally folded myelin outfoldings and has been shown to be caused by mutations in the myotubularin related protein-2 gene (MTMR2) on chromosome 11q22. Although the presence of focally folded myelin is very useful in suggesting this diagnosis, it is not specific and has been described in other forms of CMT including AR-CMT1 B2, another autosomal recessive form of CMT1.
AR-CMT1 D (CMT4D/HMSNL) is an autosomal recessive demyelinating neuropathy with a high incidence of deafness and with unusual curvilinear intra-axonal inclusions on ultrastructural examination of sural nerves. The disease was originally described in Bulgarian gypsies and since then described in other gypsy families, and is caused by mutations in the N-myc downstream regulated gene 1 (NDRG1) on chromosome 8q24.
The most recently described gene to cause AR-CMT1 is periaxin on chromosome 19q13.1-13.3, mutations of which give rise to AR-CMT1 F. A recent report has suggested that there is a higher incidence of sensory involvement with periaxin mutations than seen in other forms of autosomal recessive CMT1, which may a useful pointer in diagnosis.
Axonal CMT
CMT2 (axonal CMT) is diagnosed when the median motor nerve conduction velocity is above 38 m/s. There are autosomal dominant, autosomal recessive, and X linked forms of CMT2.
Autosomal dominant CMT2
Autosomal dominant CMT2 is reported to be less common than autosomal dominant CMT1, but as most of the genes for autosomal dominant CMT2 are unknown it is difficult to get accurate prevalence figures. It remains possible that many patients with idiopathic axonal neuropathy will eventually be shown to have CMT2. Autosomal dominant CMT2 is clinically indistinguishable from CMT1, but the motor nerve conduction velocities should be normal or near normal. Patients may present later. CMT2 is genetically heterogeneous and until recently there were no causative genes described. There are now six loci (excluding the rare cases of CMT2 caused by P0 mutations and HMSNP, which unlike classical CMT2 has pronounced proximal involvement) and two genes described (table 2).
The first gene to be described was in a Russian family with CMT2 who were found to have a mutation in neurofilament light gene (NF-L) on chromosome 8p21. Rare other families have now been described with mutations in this gene. Interestingly, in some of the other families, patients have been described with median motor nerve conduction velocities well below 38 m/s. There is still debate as to whether the low nerve conduction velocities reflect very low motor amplitudes or whether CMT secondary to NF-L mutations should be classified as intermediate CMT (see below).
The most recently described CMT2 gene is a member of the Kinesin family, microtubule motor KIF1Bβ on chromosome 1p35-p36. A mutation in this gene has been described in one family with CMT2A.
X linked CMT2
One X linked form of CMT2 (CMT 2X) has been described linked to Xq24-q26, but no gene is known for this.
Autosomal recessive CMT2
There are three/four loci and two genes described for autosomal recessive CMT2 (AR-CMT2) which appears to be a rare form of CMT (table 2). It is not yet clear if AR-CMT2 B is allelic to AR-CMT2 D/AR-CMT1 A.
Recently, mutations in Lamin A/C (LMNA) have been shown to be the cause of AR-CMT2 A. This finding was of particular interest as mutations in LMNA have been described in association with limb-girdle dystrophy type 1B, autosomal dominant Emery-Dreifuss muscular dystrophy, dilated cardiomyopathy type 1A, and autosomal dominant partial lipodystrophy.
GDAP1, mutations of which can cause an autosomal recessive demyelinating neuropathy (AR-CMT1 A, CMT4A) has also been described to cause an autosomal recessive axonal neuropathy (AR-CMT2 D). This is the second gene in CMT (Po being the first) that can cause both an axonal and a demyelinating neuropathy and raises obvious questions about mechanisms.
Intermediate CMT
Intermediate CMT does not traditionally appear in classifications but it is emerging as a clinical entity that can occasionally be useful. Families are being described with median motor conduction velocities ranging form 25–45 m/s, which are difficult to classify into CMT1 or CMT2. CMT X1 secondary to CX32 mutations can give motor conduction velocities in this range. As stated above mutations both in Po and in the GDAP1 can cause both demyelinating and axonal CMT and therefore can give rise to motor nerve conduction velocities in this range. It may be shown that mutations in NF-L will also fit into this category.
Two loci have recently been described to cause an autosomal dominant form of intermediate CMT, termed DI-CMT (table 2).
Approach to molecular diagnosis of CMT
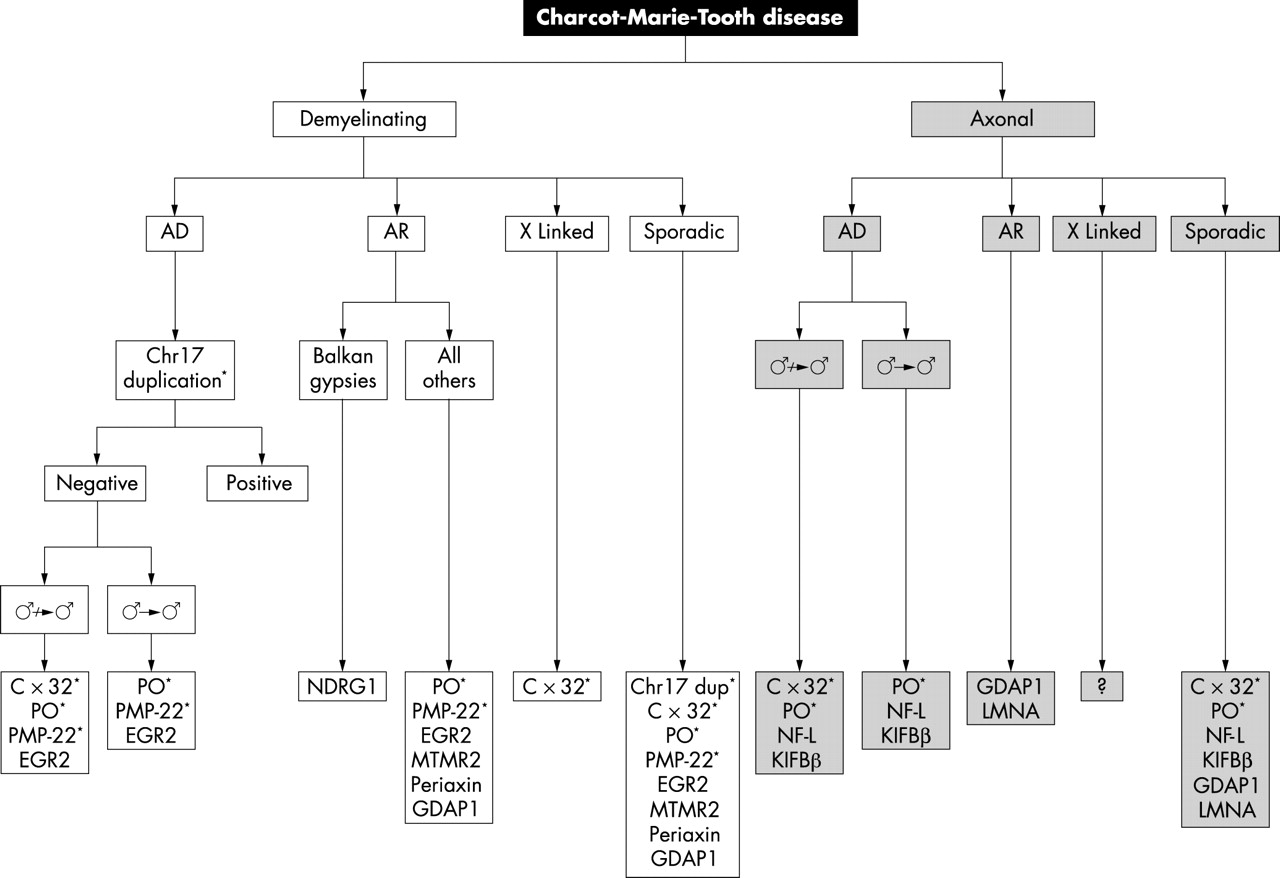
From the practising clinicians point of view, the most important questions are how to diagnose CMT, which genes should be screened for in individual cases, and, most importantly, for which genes is routine screening available in the country in which the clinician practices. Figure 1 is a flow diagram for the molecular diagnosis of CMT in the UK, highlighting those genes for which routine testing is available. This information only applies to the UK.

{kind=link}
Flow chart for the molecular diagnosis of Charcot-Marie-Tooth (CMT) disease. *Available routinely in the UK; ♂ → ♂, male to male transmission; AD, autosomal dominant; AR, autosomal recessive; gene abbreviations are listed in table 2.
The first step in the diagnosis is for the clinician to decide if CMT is the likely diagnosis. This may be obvious if there is a family history, but often in small families patients may present as sporadic cases. Useful clues that may point to a diagnosis of CMT include a long history extending back to childhood, foot deformities such as pes cavus, and in the case of demyelinating neuropathies, a symmetrical reduction in motor conduction velocities without conduction block in the right clinical context may suggest CMT rather than acquired demyelinating neuropathies such as chronic inflammatory demyelinating polyneuropathy (CIDP). If a family history is available or consanguinity is present in the family it may be possible to classify a family into autosomal dominant, autosomal recessive or in the absence of male to male transmission, probable X linked inheritance, but often the families are too small to make this possible. Nerve conduction studies are very useful and should be the first investigation used to classify the patient as having demyelinating, axonal or intermediate CMT. If there are other affected family members available, nerve conduction studies in them can help establish the probable diagnosis in the index case (for example, demyelinating motor conduction velocities in a male family member and axonal motor conduction velocities in a female family member may suggest CMT X1 secondary to CX32 mutations). Although a nerve biopsy is not usually required in the autosomal dominant and X linked cases, it still may be very useful in the autosomal recessive and sporadic cases.
After the patient has been classified clinically, neurophysiologically, and in selected cases pathologically, molecular diagnosis should be sought. These tests are often expensive and time consuming and the more directed the request by the clinician, the more cost effective and useful molecular diagnosis will be. Most genetic laboratories in the UK offer screening for the chromosome 17 duplication and deletion. Specialised genetic laboratories offer PMP-22, P0, and CX32 point mutation screening, but it is worth checking how the screening is done as some screening methods only detect about 90% of point mutations. In those genes that routine testing is available, testing can be used for diagnostic, predictive, and pre-natal tests. Screening of new genes such as EGR2, MTMR2, NDRG1, periaxin, KIFIBβ, NF-L, GDAP1, and LMNA are as yet only available in laboratories with a research interest in CMT and therefore will not currently be available to most practising clinicians.
As shown in fig 1, in a patient with a clearly demyelinating neuropathy, either dominant or sporadic and even if there is no evidence of male to male transmission, the chromosome 17 duplication should initially be screened. Following this, cases (not suggestive of DSD or CHN) with no male to male transmission should be screened for CX32 mutations. In all other demyelinating cases P0 and PMP-22 should then be screened followed lastly by EGR2 if available. In the remaining demyelinating cases, especially the more severe ones and the cases suggestive of autosomal recessive transmission, MTMR2, periaxin, and GDAP1 should be screened if available and if CX32, P0, and PMP-22 screening are negative. NDRG1 is only worth screening in patients of Balkan gypsy origin.
In the axonal form of CMT, most of the causative genes have not been identified. In autosomal dominant and sporadic cases, if there is no male to male transmission and especially if the index case is female, CX32 should be screened first. If this is negative and in all other cases, P0 should be screened next. If these are both negative, and they will be in most cases, it is worth checking NF-L and KIFIBβ where available. Most axonal cases will currently remain undiagnosed at the molecular level. In axonal cases where the inheritance is clearly recessive GDAP1 and LMNA should be screened. There are no genetic tests available for X linked CMT2.
Finally, in the intermediate cases, although they are not shown as a separate entity in the flow chart (fig 1), it is occasionally useful to consider them separately. In these cases, it is still recommended to check the chromosome 17 duplication first. If this is negative and there is no male to male transmission, we suggest checking CX32 next and then in all remaining negative cases checking P0 and GDAP1 if available. In intermediate cases that are still negative, especially if the motor nerve conduction velocities are below 38 m/s in the median nerve, it is worth checking PMP-22 and EGR2 for point mutations. It remains to be seen how useful routine screening of the more recently described rare genes causing CMT will be in intermediate cases.
Hereditary sensory and autonomic neuropathies
The hereditary sensory and autonomic neuropathies (HSAN),5 also called the hereditary sensory neuropathies (HSN), are rarer than CMT, but there have been major advances in the identification of the causative genes in the last decade. HSAN is classified into five types although type 4 and 5 may be allelic and three causative genes have been identified (table 3).
Hereditary sensory and autonomic neuropathies
HSANI is an autosomal dominant disorder presenting in the second or third decade and characterised by a progressive degeneration of the dorsal root ganglia and motor neurons leading to distal sensory loss and later muscle wasting and weakness. Sensory involvement is predominant and can lead to complications such as ulceration and amputations. Many families with HSAN1 (but not all) have been found to have mutations in the serine palmitoyltransferase, long chain base subunit-1 (SPTLC1) gene on chromosome 9q. There is a high incidence of distal lancinating pains in these patients which can be useful diagnostically as positive sensory symptoms are rare in other inherited neuropathies such as CMT.
HSANII is an autosomal recessive neuropathy characterised by an early onset sensory neuropathy affecting all modalities of sensation in the upper limbs, lower limbs, and trunk. No genetic loci or genes have been described for this type of HSAN.
HSANIII (Riley-Day syndrome, familial dysautomonia) is another autosomal recessive neuropathy mainly seen in Askenazi Jewish families. Clinically, autonomic symptoms predominate but the peripheral sensory and motor neurons are also involved. HSANIII has been shown to be caused by mutations in the gene encoding the IκB kinase complex-associated protein (IKAP).
HSANIV is a rare autosomal recessive neuropathy characterised by congenital insensitivity to pain and anhidrosis (CIPA). The cardinal features are recurrent episodes of unexplained fever, anhidrosis, absence of reactions to noxious stimuli, self mutilating behaviour, and mental retardation with an almost complete loss of unmyelinated fibres on nerve biopsy. This disease is caused by mutations in the tyrosine kinase A receptor (TRKA) for nerve growth factor.
HSANV, an autosomal recessive sensory neuropathy, is similar to HSANIV but is characterised by a selective loss of small myelinated fibres. One family with HSANV has also been shown to have mutations in TRKA so these two conditions may be allelic.
None of the above HSAN genes are available for routine testing in the UK, but some are done in laboratories with a research interest in HSAN.
GENETIC MUSCLE DISEASES
Muscular dystrophies
Many genes causing different muscular dystrophies have been discovered in recent years. The causative proteins have various functions indicating that dystrophic muscle may be the relatively non-specific end result of a number of different pathogenic pathways. The proteins identified to date include sarcolemmal structural proteins (for example, dystrophin, sarcoglycans, dysferlin), nuclear envelope proteins (emerin, lamin a/c), enzymes (calpain, Fukutin related protein), sarcomeric proteins (myotilin), and extracellular matrix proteins (laminin, collagen type 6). For some disorders, such as myotonic dystrophy, a single gene accounts for most cases and a simple gene test is available. In contrast, for other disorders, such as the limb-girdle muscular dystrophies, the situation regarding testing is more complex because of genetic heterogeneity.
Xp21 dystrophies; dystrophinopathies
-
▸ DNA based diagnosis available?
Yes, but negative gene test does not exclude diagnosis
-
▸ Muscle biopsy needed?
Yes, if genetic test negative
Xp21 dystrophies: the dystrophinopathies
Dystrophin is a major muscle cell structural protein which lies just below the sarcolemma. It has an important role in stabilising the sarcolemma during muscle contraction. It links intracellular actin to a complex set of transmembrane proteins which in turn link to elements of the extracellular matrix (particularly to an extracellular protein called laminin). Genetic defects in the dystrophin protein are associated with two major phenotypes: Duchenne, and Becker muscular dystrophy.
Duchenne is an aggressive and lethal dystrophy affecting boys. It associates with a severe reduction in the amount of the dystrophin protein in muscle. To date there is no curative treatment, but improved ventilatory support has recently been shown to increase substantially the quality of life and life expectancy in such boys.
Becker muscular dystrophy is more likely to be encountered in adult muscle practice. It varies widely in clinical severity and there is a correlation with the amount of dystrophin protein. Generally, Becker patients present as a limb-girdle pattern of muscle weakness with pseudohypertrophy of calf muscles being a common feature. Cardiomyopathy is an important complication of dystrophinopathies and occasionally a severe dilated cardiomyopathy can be the presenting feature.
Diagnosis can be achieved by genetic testing on a blood sample. However, the dystrophin gene is very large and most testing protocols only screen a proportion of the gene. Therefore a “negative” genetic test does not exclude the diagnosis and if appropriate a muscle biopsy with dystrophin immunological studies is required.
Genetic counselling in families as for an X linked recessive disease is important. Female carrier status may be ascertained by dosage sensitive polymerase chain reaction (PCR). However, it is notable that approximately one third of dystrophinopathy cases represent new mutations without preceding family history. This emphasises the importance of the need for an effective treatment.
Limb-girdle muscular dystrophy
There have been many recent gene discoveries in limb-girdle muscular dystrophy (LGMD). A surprising array of proteins with different functions have been shown to cause the LGMD phenotype (table 4). Identified proteins include muscle enzymes and nuclear envelope proteins as well as sarcolemmal structural proteins. Although patients generally have the common feature of a limb girdle pattern of muscular weakness without facial involvement, clinical differences exist. For example, the propensity to develop cardiomyopathy or respiratory muscle failure varies considerably in different forms. Accurate DNA based diagnosis will therefore allow logical monitoring programmes of cardiac and respiratory function to be developed. Autosomal recessive LGMD is far more common than the dominant form. Many of the initial genes discovered are mainly associated with a paediatric age onset, but recent discoveries have identified forms which may present in adult life. Of particular note is the recent identification of mutations in the gene known as the Fukutin related protein (FKRP). FKRP codes for a glycosylation enzyme and recent data suggest it may be the most common cause of adult onset LGMD identified to date. A predominant mutation (C826A) seems to account for many cases which should make DNA based diagnosis easier. In general, patients with recessive LGMD should be investigated initially by immunological studies on skeletal muscle. This will usually identify a reduction in the causative protein. The appropriate gene can then be analysed to establish a genetic diagnosis.
LGMD
-
▸ DNA based diagnosis available?
Yes, but only after muscle protein immunochemistry has reduced number of candidate proteins via National Specialist Commissioning Advisory Group in UK
-
▸ Muscle biopsy needed?
Yes, muscle biopsy remains mandatory
Emery Dreifuss muscular dystrophy
Emery Dreifuss muscular dystrophy (EDMD) is a readily recognisable muscular dystrophy which should be considered if early contractures is a prominent clinical feature. Patients present with a scapulohumeroperoneal pattern of muscular weakness and often have strikingly thin muscles. Contractures of the cervical extensor muscles as well as the biceps and long finger flexor tendons are a common early feature. Cardiac conduction defects are common and cardiac screening is important.
EDMD is now known to be caused by mutations in genes coding for proteins located in the membrane surrounding the muscle cell nucleus. The exact function of such nuclear envelope proteins is uncertain but they may have a role in maintaining the integrity of the nuclear membrane or in controlling release of nuclear mRNA
The nuclear proteins include emerin, which causes the X linked form of EDMD, and lamin A/C, which causes the less common dominant and recessive forms. An accurate genetic diagnosis is therefore critical in order to provide accurate genetic counselling. Mutations in the lamin A/C gene have also been described in a form of LGMD (LGMD1B, table 4) in an autosomal recessive axonal neuropathy and in the skin disorder partial lipodystrophy.
EDMD
-
▸ DNA based diagnosis available?
Yes, but usually only after muscle protein immunocytochemistry (for lamin A/C and emerin)
-
▸ Muscle biopsy needed
Yes, muscle biopsy remains important
Myotonic dystrophy and proximal myotonic myopathy
Myotonic dystrophy (MD) is an important autosomal dominant multisystem disorder. It ranges in severity from a severe congenital myopathy, which may be fatal, to late onset isolated cataracts. Neuromuscular symptoms in a typical case include facial weakness, mild distal myopathy, and myotonia. Variably present additional symptoms include cataracts, endocrine disturbance such as diabetes mellitus, cardiomyopathy, hair loss, cognitive slowing, daytime somnolence, bowel dysmotility, and respiratory muscle weakness. It is the presence of one or more of these extra muscle features in combination with typical muscle symptoms which usually leads to the correct diagnosis on clinical grounds. However, diagnostic difficulty may arise if the muscular symptoms are not evident or are subtle. For example, we have seen cases presenting initially to diabetic, cardiac, gastrointestinal or memory clinics before full diagnosis.
Anticipation, the increased clinical severity of a genetic disease in succeeding generations, is recognised in myotonic dystrophy. The molecular correlate of anticipation has been shown to be an unstable trinucleotide repeat (CTG) in the 3 prime untranslated region of a gene on chromosome 19 called myotonin kinase. Recent evidence suggests that the expanded repeat may exert its toxic affect at the RNA level. There is evidence that the expanded untranslated repeated sequence in RNA forms abnormal degradation resistant nuclear deposits (aggregates). Such nuclear deposits probably impair nuclear matrix function resulting in altered expression of other genes. This effect on other genes may, in part, explain the diverse multi-system clinical involvement. For example, there is evidence that expression of the skeletal muscle voltage gated chloride channel is altered in MD patients. The resulting chloride channel dysfunction is the probable basis of the myotonia. This is of interest since it is recognised that primary mutations in this chloride channel are the basis of the separate pure myotonic disorder myotonia congenita.
The diagnosis of myotonic dystrophy is usually straightforward on clinical grounds with the above exceptions noted. DNA based diagnosis is the initial investigation of choice and muscle biopsy is not usually needed. A detailed family history will usually reveal other affected members and an autosomal dominant pedigree should be constructed. Genetic counselling should be as for an autosomal dominant disorder with full penetrance but variable expression. It is particularly important to identify females at risk of inheriting the gene in pedigrees. This is because the severe, often lethal, congenital form is confined to the offspring of affected females. Such females can then be offered appropriate genetic counselling and also prenatal diagnosis if they wish. Patients with myotonic dystrophy should be followed up in a muscle clinic where they can be monitored for systemic complications. There is currently no curative treatment, but the potential now exists to develop drugs which may reduce the abnormal RNA deposition. In most patients myotonia is not symptomatic and does not need treatment. In those cases with symptomatic myotonia we suggest mexilitene is the drug of choice provided the cardiac QT interval is normal.
Myotonic dystrophy and PROMM
-
▸ DNA based diagnosis available?
Yes, through regional clinical genetics laboratory
-
▸ Muscle biopsy needed?
Not usually, unless the genetic tests for MD and PROMM are negative
If the genetic test for MD is negative, consideration should be given to a recently recognised disorder known as PROMM (proximal myotonic myopathy). This less common disorder is characterised by proximal rather than distal myopathy, but in other respects may exhibit similar features to MD. Muscle pain may be a prominent feature which may suggest the diagnosis on clinical grounds. The liver γ-GT enzyme concentration may be raised. The genetic defect underlying this autosomal dominant disorder has recently been identified as a CCTG repeat sequence in intron 1 of a zinc finger protein gene on chromosome 3. As in MD there is evidence that this untranslated defect operates at the RNA level, resulting in nuclear aggregates. Mild anticipation is reported in PROMM families but a severe congenital form has not been described to date.
Fascioscapulohumeral muscular dystrophy
Fascioscapulohumeral muscular dystrophy (FSHD) is an important autosomal dominant disorder frequently encountered in adult specialist muscle practice. Patients commonly have facial muscle weakness as the initial symptom, which may be followed by periscapular and humeral muscle involvement. Later in the disease course lower limb weakness, particularly anterior tibial, and abdominal wall weakness may occur. It is notable that the distribution of weakness is often asymmetric, with the right side often being more involved. Isolated periscapular weakness without facial involvement may occur. The severity is variable ranging from isolated asymmetrical scapular winging through to pronounced early onset weakness rendering the patient wheelchair bound. Scoliosis, deafness, and retinal vasculopathy are uncommon additional features.
FSHD is almost fully penetrant by the age of 30 years. It has been known for over 10 years that the gene for FSHD is located on chromosome 4, but identification of the exact gene has remained elusive. Surprisingly, a search of the mapped region did not reveal any genes but did reveal contraction (truncation) of a normally occurring non-coding repeated DNA sequence. The presence of this truncated region of DNA has been shown to associate strongly with chromosome 4 linked FSHD and this is the basis of the available genetic test. It is important to note that this is an indirect genetic test in that, unlike all the other gene tests described in this article, it is not the gene itself which is being analysed. Nevertheless, this truncation detection genetic test is a useful aid in the precise diagnosis of FSHD. It is believed that truncation of this region may have a detrimental effect on the true FSHD gene which is suspected to be located elsewhere on chromosome 4—known as the positional variegation effect.
FSHD
-
▸ DNA based diagnosis available?
Yes, indirect genetic test available through regional genetics laboratory
-
▸ Muscle biopsy needed?
Not usually unless genetic test for FSHD negative
Inflammatory muscle biopsy may cause difficulty distinguishing from polymyositis
OPMD
-
▸ DNA based diagnosis available?
Yes
-
▸ Muscle biopsy needed?
Not usually
Oculopharyngeal muscular dystrophy
Oculopharyngeal muscular dystrophy (OPMD) is an uncommon disorder characterised by late onset pharyngeal muscle weakness and ophthalmoplegia. It is usually a highly penetrant dominant disorder although recessive inheritance may occur. OPMD may enter into the differential diagnosis in patients with late onset ophthalmoplegia. The most common genetic defect is an expansion of a short [GCG]7–13 triplet in the polyAbinding protein 2 (PAB2) gene on chromosome 14q. Genetic testing for the expanded repeat sequence is available and usually obviates the need for muscle biopsy.
Muscle ion channel disorders
Periodic paralyses
The periodic paralyses (PP) are autosomal dominant disorders in which patients experience attacks of focal or generalised muscle weakness usually beginning in the first or second decade. The onset of such attacks is usually accompanied by a change in the serum potassium concentration and the disorders have been divided into two predominant forms; hyperkalaemic periodic paralysis, and hypokalaemic periodic paralysis. These disorders are caused by mutations in voltage gated ion channel genes and the resulting disturbed channel function results in altered membrane excitability which is the basis of the attacks. Hyperkalaemic PP associates with mutations in the skeletal muscle voltage gated sodium channel gene SCN4A. The more common hypokalaemic form of PP associates with mutations in a voltage gated calcium channel CACNA1S. Vigorous exercise followed by rest is a potent stimulus to attacks in both forms of PP. In both forms attack frequency is reduced by acetazolamide or dichlorphenamide. Typically the attack frequency declines with age and many patients develop a fixed vacuolar myopathy which can be significantly disabling. The diagnosis can usually be confirmed by a combination of electrophysiology (particularly the short exercise test) and DNA testing. Provocative tests are usually not required and muscle biopsy may be normal or only non-specifically myopathic. Recently, the genetic basis of Andersons syndrome (periodic paralysis, facial dysmorphism, and malignant cardiac arrhythmias) has been shown to be in a potassium channel gene (KCNJ2). It is important to consider this diagnosis in patients with PP who have an abnormal ECG or a history of cardiac arrhythmias.
Periodic paralysis
-
▸ DNA based diagnosis available?
Yes
-
▸ Are provocative tests needed?
Not usually
-
▸ Muscle biopsy needed?
Not usually
Inherited myotonias
Paramyotonia congenita, potassium aggravated myotonia, and myotonia congenita are all genetic myotonic disorders caused by mutations in voltage gated ion channels. Paramyotonia congenita associates with point mutations in the muscle sodium channel SCN4A. Patients experience increasing myotonia (muscle stiffness) the more a muscle is contracted. The face and upper limbs are particularly affected. The activity induced muscle stiffness is often further exacerbated by cold temperatures.
Potassium aggravated myotonia (now also known as sodium channel myotonia and encompassing myotonia fluctuans, myotonia permanens, and acetazolamide responsive myotonia) is a rare pure myotonic disorder of variable severity. Some patients report striking daily fluctuations in the severity of the myotonia and others note cold worsening. Striking worsening of the myotonia on potassium ingestion may occur.
Myotonia congenita is the most common pure myotonic disorder. It is caused by mutations in the voltage gated chloride channel (CLCN-1) on chromosome 7q. Dominant and recessive forms exist. Patients exhibit generalised myotonia which is worse after a period of rest but improves with repeated contractions—“warm-up phenomenon”. Patients with myotonia congenita are often significantly symptomatic from the myotonia and in our experience mexilitine is the drug of choice.
Inherited myotonias
-
▸ DNA based diagnosis available?
Yes
-
▸ Muscle biopsy needed?
Not usually
Metabolic muscle disorders
Mitochondrial respiratory chain diseases
A bewildering array of clinical phenotypes may associate with dysfunction of the mitochondrial respiratory chain. Fortunately, the most common mitochondrial phenotypes presenting to the adult neurologist are readily recognisable. A combination of genetic and muscle histochemical and biochemical investigations can usually result in an accurate diagnosis. Mitochondrial respiratory disease is quite common. One recent study suggests that in a UK population 16 in 100 000 either have or are at risk of mitochondrial disease.
Mitochondrial respiratory chain diseases
-
▸ DNA based diagnosis available?
Yes but there may be an age associated decline in MtDNA mutation load in blood
-
▸ Muscle biopsy needed?
Yes, if blood MtDNA analysis negative
Muscle needed for mitochondrial enzyme analysis
Since normal function of the respiratory chain is dependent on the interplay of nuclear and mitochondrial genes, it follows that respiratory chain diseases may be inherited in either an autosomal or maternal pattern.
The majority of adult mitochondrial phenotypes associate with primary mutations in mitochondrial DNA (MtDNA) (table 5). These include large scale rearrangements (such as deletions) which are generally sporadic and not passed on to the succeeding generation. For example, if a deletion is identified in a patient with CPEO it is appropriate to counsel on the basis that this will not be passed on. In contrast the other common mutations in MtDNA, point mutations, are usually passed down the maternal line. Unfortunately, the factors which determine the amount of a particular point mutation which may be passed down cannot be predicted with accuracy. Thus, it is extremely difficult to offer women who harbour disease causing point mutations accurate recurrence risks. Until current research efforts identify the factors which influence the recurrence risks for point mutations, the only definite way to avoid maternal transmission is to consider ovum donation.
Common mitochondrial phenotypes in adults
Glycogenoses and lipid storage disorders
Disorders of muscle energy production include the glycogenoses and lipid storage disorders. Patients with these recessive disorders generally develop muscle pain on exertion. Myoglobinuria and sometimes fulminant rhabdomyolysis may occur.
Patients with McCardle’s disease (the most common glycogenosis caused by myophosphorylase deficiency) present with muscle pain soon after commencing exercise. The pain can be severe and cramping, often causing the patient to stop. A second wind phenomenon is common. The forearm non-ischaemic lactate test will show a blunting of the expected rise in muscle lactate induced by repeated isometric exercise because of a failure to breakdown glycogen. Muscle biopsy will allow histioenzymatic staining for myophosphorylase. Genetic testing is available but is usually done after the metabolic defect has been confirmed.
Of the many disorders of lipid metabolism, the most common lipid storage disorder is carnitine palmitoyl transferase-II (CPT-II) deficiency. Adults with CPT-II deficiency usually present with muscle pain induced by prolonged exercise, and often the pain may not develop until some time after exercise. Myoglobinuria post-exercise is frequent. Rhabdomyolysis is more likely to occur after fasting or if there is intercurrent infection or dehydration. Hypoketotic hypoglycaemia may occur. Occasionally patients with CPT-II deficiency present with a painless proximal myopathy. Muscle biopsy light microscopy may reveal abnormal accumulations of lipid. Muscle tissue can be used for specific enzyme assays. The pattern of acylcarnitines in blood is particularly useful whenever a lipid storage disorder is being considered. Molecular genetic diagnosis is available.
Metabolic muscle disorders (glycogenoses, lipidoses)
-
▸ DNA based diagnosis available?
Yes
-
▸ Specialist metabolic testing?
Yes, very useful
Non-ischaemic lactate test (glycogenoses)
Blood acylcarnitine/urinary organic acids (lipid disorders)
-
▸ Muscle biopsy needed
Yes
Useful sources of information on neuromuscular diseases
-
▸ More information about the UK, Department of Health funded, National Advisory and Diagnostic Service for Rare Neuromuscular Disorders (NSCAG) is available from Dr MG Hanna: mhanna{at}ion.ucl.ac.uk
Patient support groups
-
▸ Muscular Dystrophy Campaign UK: www.muscular-dystrophy.org/
-
▸ The Neuropathy Trust: www.neuropathy-trust.org
-
▸ Charcot-Marie-Tooth Society UK: www.cmt.org.uk
CONCLUSIONS
The diagnosis of patients with neuromuscular disorders has truly entered the molecular genetic era. DNA based diagnosis is now available for many of these disorders and often removes the need for invasive tests such as muscle or nerve biopsy. Efficient selection of genetic tests is important and neurophysiology has a particularly important role in this regard for genetic neuropathies and muscle channelopathies. Muscle biopsy remains essential in certain situations—for example, in the comprehensive investigation of patients with limb-girdle muscular dystrophies and in some metabolic myopathies. It is worth noting that while a blood sample is all that is required for testing in nuclear gene disorders, muscle tissue remains most useful for mitochondrial DNA analysis.
In our view all clinical neurologists need to be aware that a wealth of genetic diagnostic possibilities exists for these patients, and that achieving such diagnostic precision on a routine basis is important. Specialist neuromuscular neurologists (ideally one for muscle and one for nerve) must exist in each region in the UK. Such specialists will be knowledgeable about the details of the genetic testing which is available. Furthermore, they will establish the appropriate infrastructure in collaboration with clinical and laboratory geneticists. In the UK we need to get on and arrange such systems now so that genetically defined patients can take full advantage of the therapies which are likely to arrive before long.
Acknowledgments
Our research is supported by the MRC cooperative “Mitochondria in health and disease”, the Wellcome Trust and the Special Trustees of the Middlesex Hospital. Our clinical muscle channel service is supported by the National Specialist Commissioning Advisory Group, Department of Health, UK. We thank Dr Nick Davies for helpful comments on the manuscript.
Abbreviations
CIDP: chronic inflammatory demyelinating polyneuropathy
CIPA: congenital insensitivity to pain and anhidrosis
CHN: congenital hypomyelinating neuropathy
CMT: Charcot-Marie-Tooth
CPT-II: carnitine palmitoyl transferase-II
DSD: Dejerine Sottas disease
EDMD: Emery Dreifuss muscular dystrophy
FKRP: Fukutin related protein
FSHD: fascioscapulohumeral muscular dystrophy
LGMD: limb-girdle muscular dystrophy
HMSN: hereditary motor and sensory neuropathies
HNPP: hereditary neuropathy with liability to pressure palsies
HSAN: hereditary sensory and autonomic neuropathies
HMN: hereditary motor neuropathy
MD: myotonic dystrophy
MtDNA: mitochondrial DNA
OPMD: oculopharyngeal muscular dystrophy
PP: periodic paralysis
PROMM: proximal myotonic myopathy