Article Text

Statistics from Altmetric.com
Introduction
Patients with endocrine dysfunction rarely seek advice from neurology. However, patients with parasellar disease may be referred because of visual loss, visual field defects, ophthalmoplegia, headache or seizures, each of which may result from compression of parasellar structures by a parasellar mass. Many neoplastic, vascular and inflammatory processes may originate from any of the pituitary, neural, vascular, dural, bony tissues in the area and also the cerebrospinal fluid (CSF).
MRI is the imaging modality of choice because of its excellent soft tissue resolution, the absence of artefact from adjacent bone and its triplanar imaging capability. CT may confer complementary information if there is suspected primary bony skull base pathology or possible calcification in a parasellar lesion.
Anatomy revisited
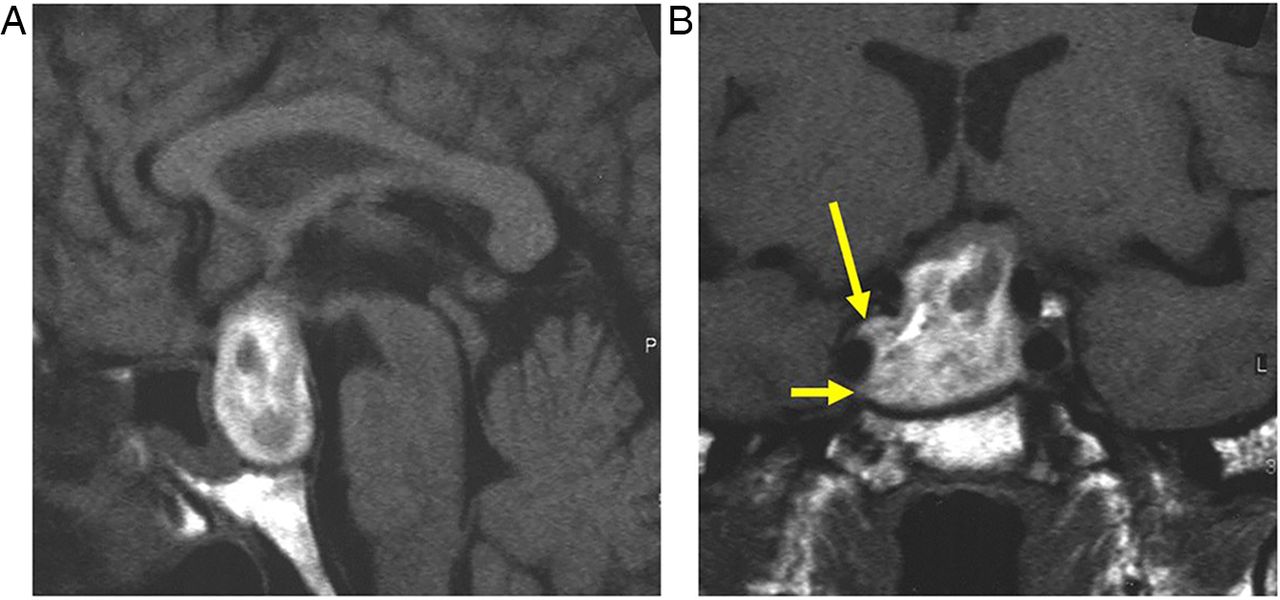
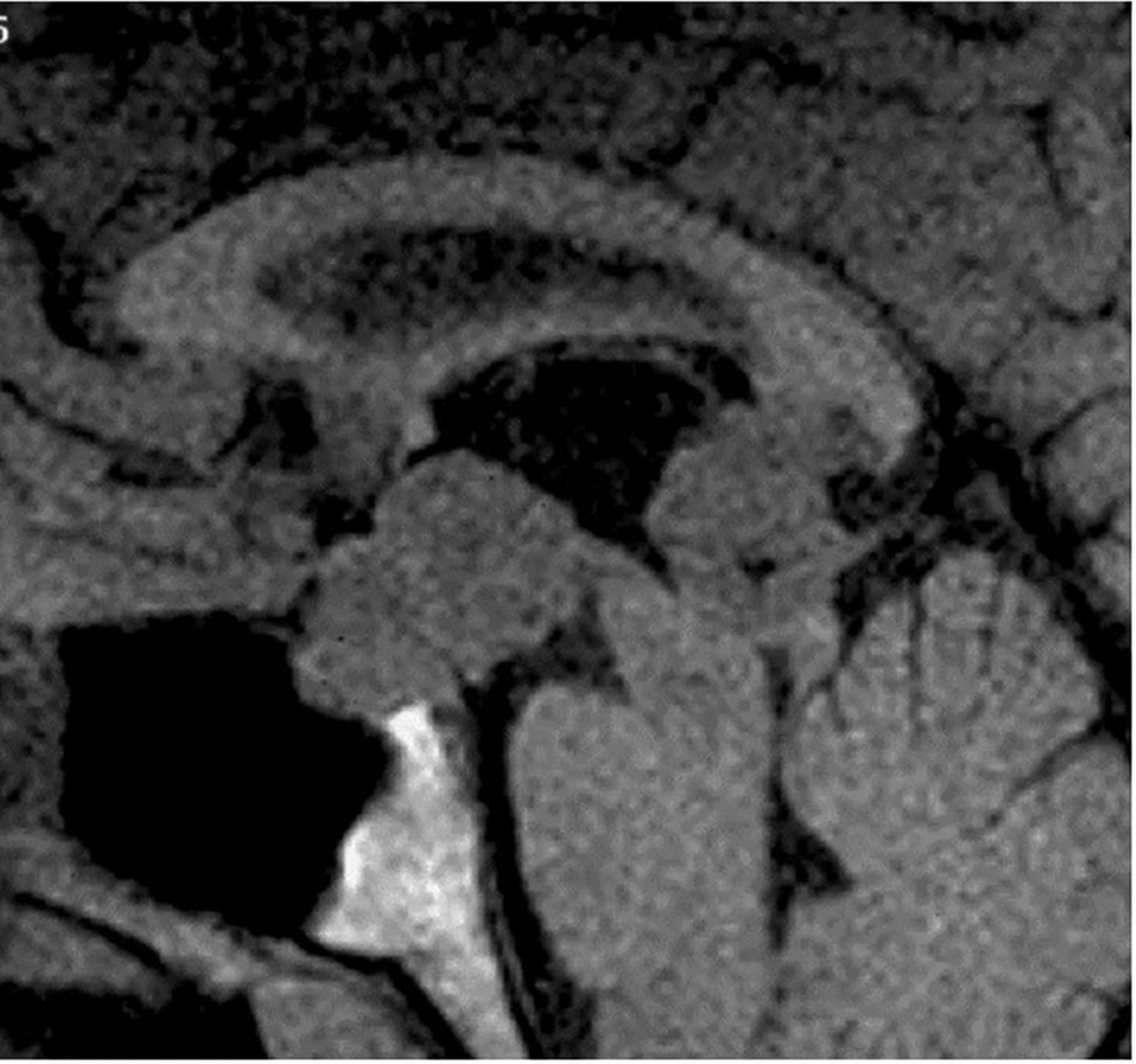
The pituitary gland sits in the sella turcica, within the central portion of the sphenoid bone and is covered by dura, the diaphragma sellae (figure 1). The cavernous sinuses (see later) lie on either side. Anteriorly is the tuberculum sella and posteriorly the dorsum sella and clivus. Inferior to the fossa is the sphenoid sinus. The optic chiasm (shaped like a figure of 8 on its side) and hypothalamus are superior in the suprasellar cistern, with the Circle of Willis and anterior end of the third ventricle. The infundibulum, tapering as it descends (3 mm wide at its hypothalamic origin and 2 mm at its insertion into the pituitary), extends from the hypothalamus, passing behind the chiasm, through the diaphragma, to the posterior pituitary. It contains fibres from the supraoptic and paraventricular nuclei and fibres, which convey the releasing hormones. The infundibulum often deviates from the midline in normal people. The posterior pituitary returns high T1 signal on MR because vasopressin-associated carrier protein (neurophysin) is a very high molecular weight glycoprotein that complexes with vasopressin to form insoluble crystal aggregates that return high signal on T1W. It is absent in central diabetes insipidus.
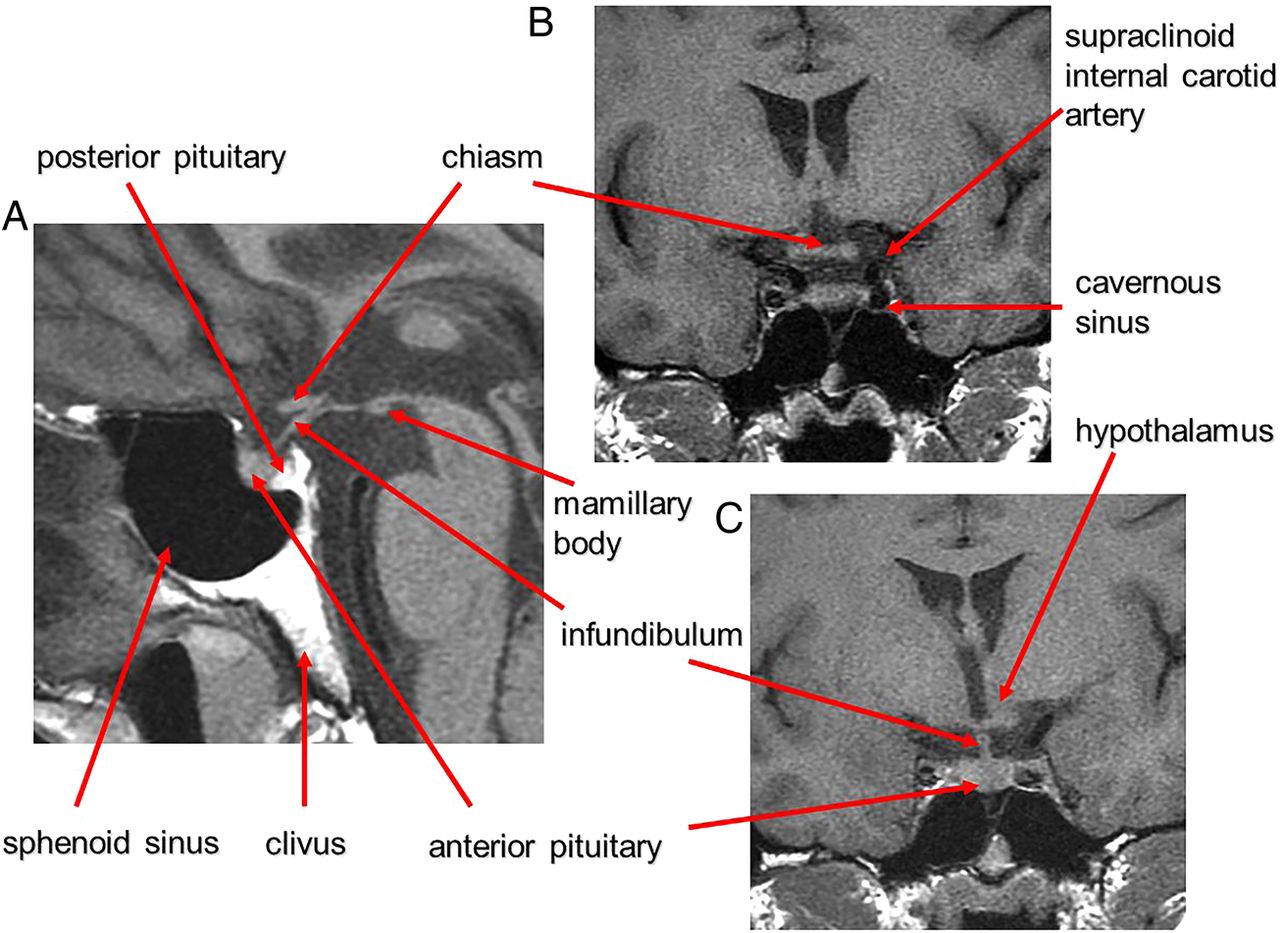
Anatomy revised. T1W midline sagittal (A) and T1W coronal (B and C) MRIs through the pituitary fossa.
The anterior pituitary has the same density as white matter on T1W MRI. It enhances with gadolinium because it lacks a blood–brain barrier, although the enhancement may be inhomogeneous.
The size and shape of the pituitary gland varies (often <6 mm height but may be >9 mm in 0.5% of healthy subjects) with sex and age, being generally larger (10 mm in height) in women. In prepubertal girls and in the peripartum period, the pituitary may appear enlarged, with a convex upper border. A bulky homogenous pituitary gland results from exogenous oestrogens, excessive production of ectopic hypothalamic-releasing factors and following venous congestion in spontaneous intracranial hypotension. A hypoplastic pituitary gland may accompany midline anomalies of the visual pathway, septum pellucidum and skull base. The anterior pituitary, infundibulum and sella are small. The posterior pituitary bright spot may be ectopic or absent. Hypoplasia usually manifests in childhood by growth hormone deficiency (pituitary dwarfism).
Pituitary incidentalomas
These are pituitary lesions (usually small, <10 mm) discovered incidentally (figure 2). They are mostly non-functioning microadenomas, occasionally Rathke's cleft cysts. Autopsy studies have found incidentalomas in 1.5–27% (mean 10.6% autopsies) of patients without pituitary disease in life. About 40% stain for prolactin, 14% for adrenocorticotropic hormone and fewer for other hormones.
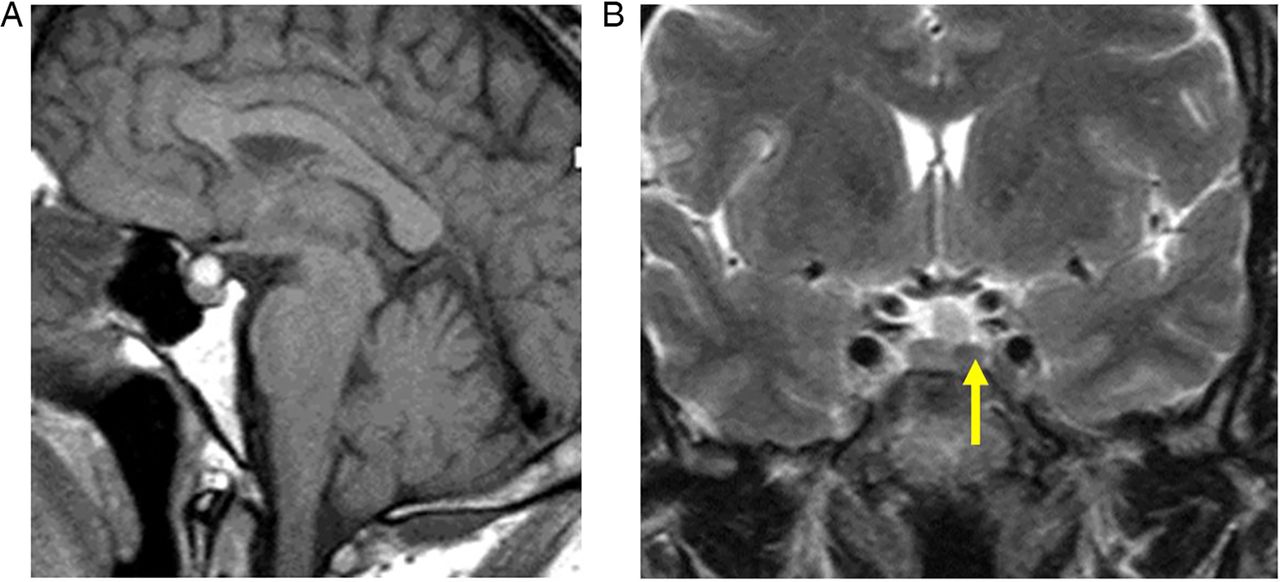
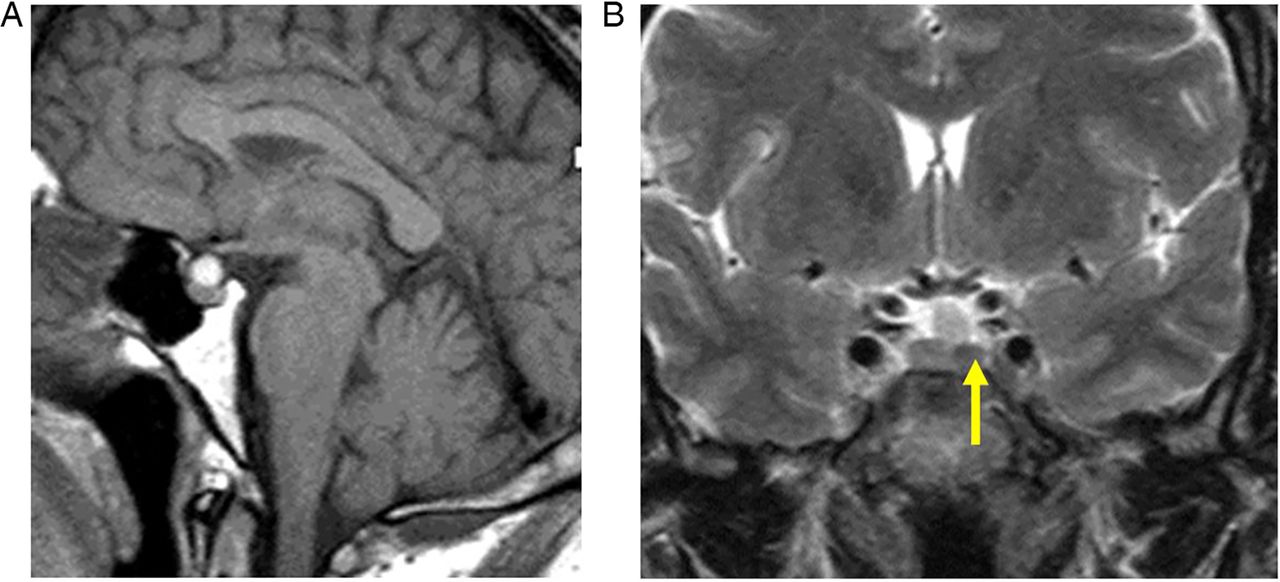
Incidentalomas. T1W sagittal (A) and T2W coronal (B) MRIs show a small slightly T2 hypointense lesion (B, arrow) in the left anterior pituitary gland—an ‘incidentaloma’. The patient had normal endocrine function. There is also a T1-hyperintense lesion adjacent to the superior aspect of the pituitary gland, likely to represent a Rathke's cleft cyst or small craniopharyngioma. The signal does not suppress with fat saturation, making a lipoma unlikely. It does not extend to the optic chiasm, and there was no visual field defect. Interval scanning confirmed stability.
These focal MR hypointensities (<10 mm diameter ‘microincidentalomas’) occur on 10–38% of MRI scans. Larger lesions (‘macroincidentalomas’) are much less common, found on 0.2–0.3% of MR scans. These patients require endocrine assessment, 6-month MR follow-up and then 12-month follow-up. Small stable lesions probably do not need further imaging. A non-functioning lesion is unlikely to turn into a functioning one. In total, 10–20% increase in size, 70–84% remain unchanged and the remainder decrease over 4–8 years.
Larger lesions require visual field assessment. Surgery may be warranted for macroincidentalomas that lie close to chiasm or cause a visual field defect.
Also, 24–50% of macroincidentalomas increase in size, 13% decrease and 63% are unchanged after a mean follow-up of 4.3 years.
Sella-based disorders
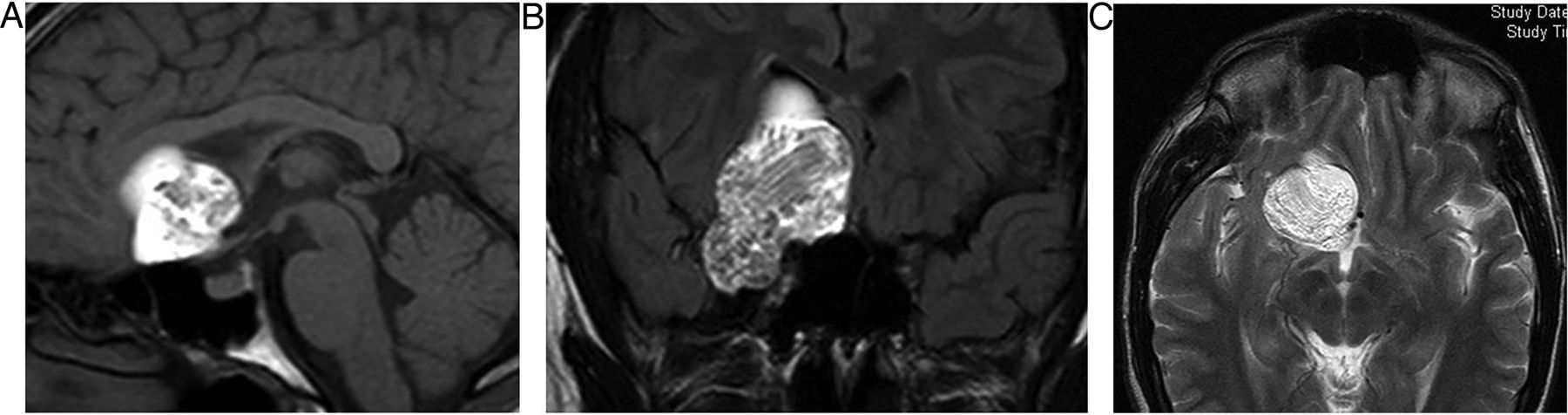
Pituitary adenomas
Adenomas account for 90% of sellar and parasellar masses. They are benign. Microadenomas are <10 mm: macroadenomas are >10 mm. Microadenomas may be non-functioning or functioning; 40% of functioning adenomas secrete prolactin.
Macroadenomas are twice as common and often present with visual loss. Infundibular distortion by a suprasellar or parasellar mass may also cause hyperprolactinaemia.
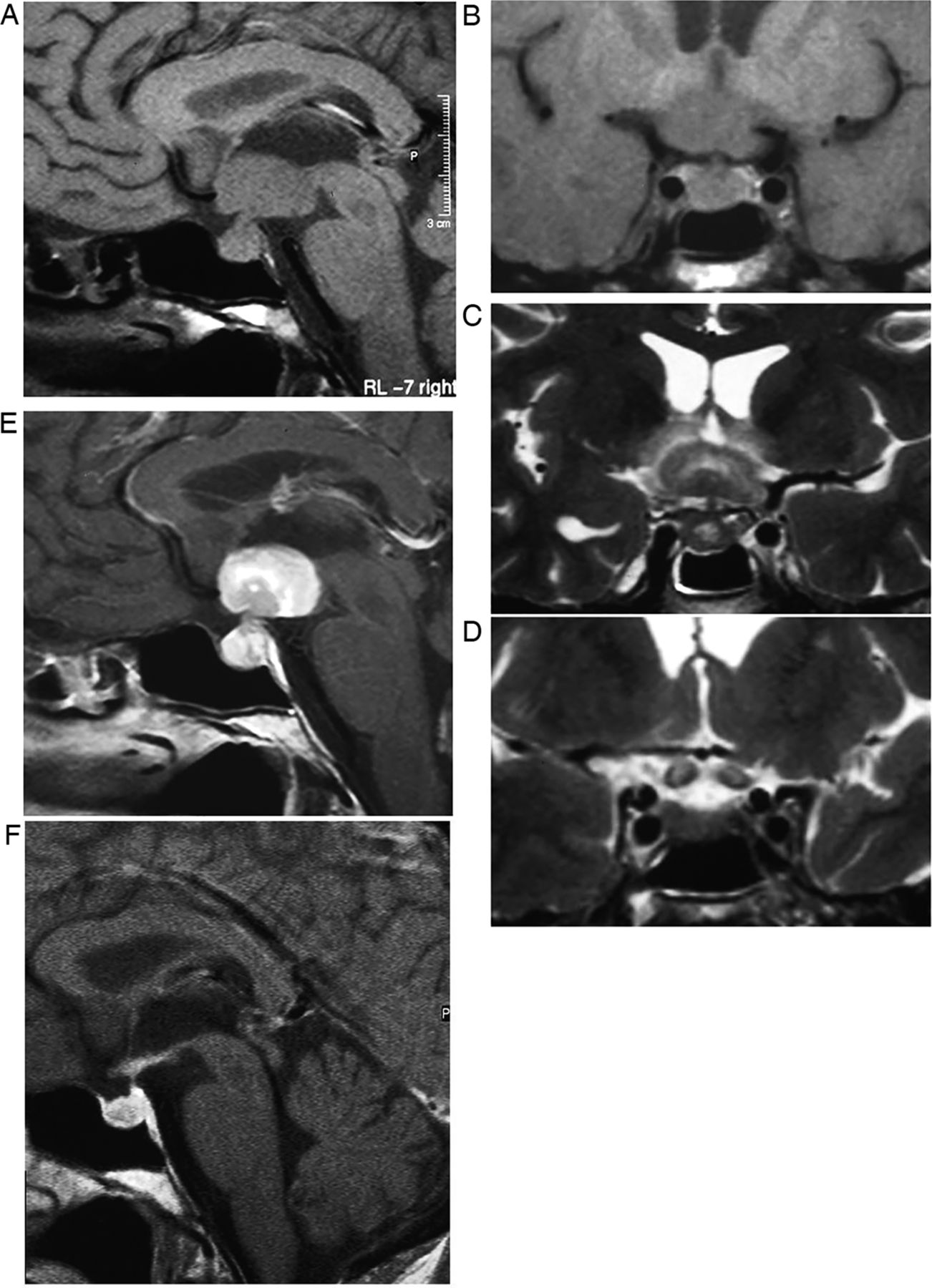
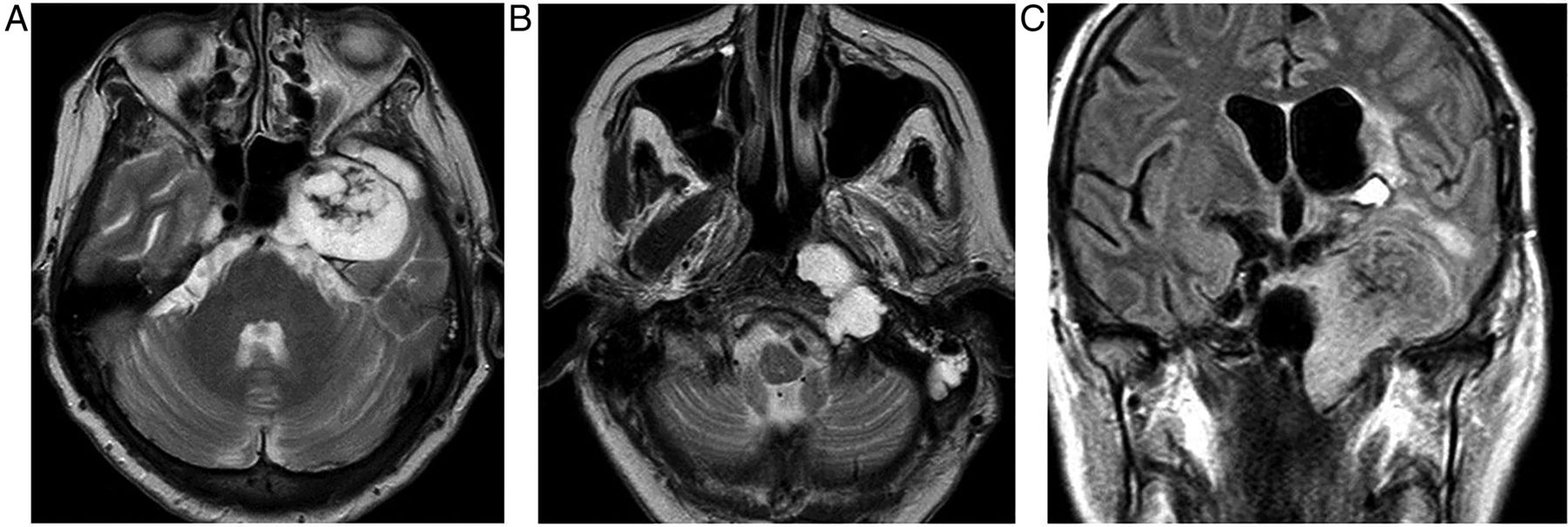
Macroadenomas (figure 3) grow slowly, expand the sella, deviate the infundibulum, produce an upward convexity to the gland and form a ‘waist’ or ‘snowman shape’ as they erode superiorly through the diaphragma into the suprasellar cistern, where they may compress the chiasm. They are T1-hypointense or isointense. The signal is variable on T2W and enhancement is homogenous or heterogeneous with cystic/necrotic foci; 10% are haemorrhagic (especially after treatment with bromocriptine).

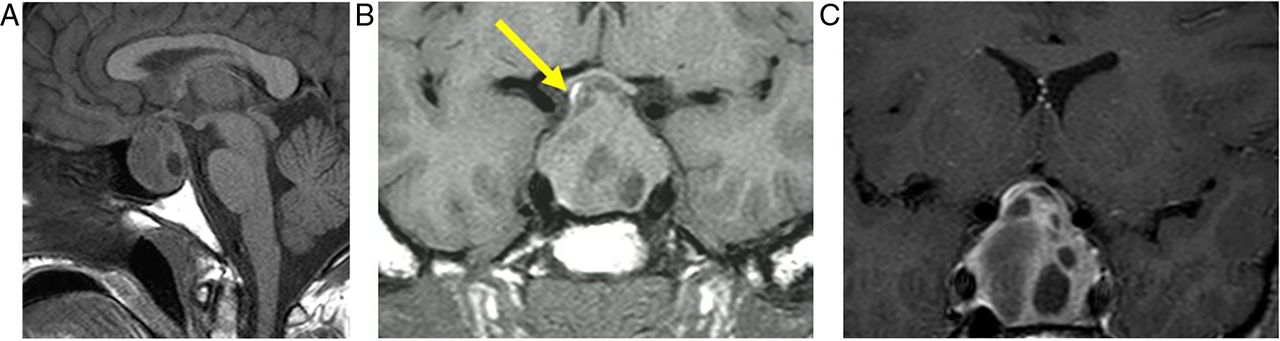
Pituitary macroadenoma: T1W sagittal (A) and coronal (B) and T1W gadolinium-enhanced coronal (C) images of the pituitary fossa show expansion of the sella by a cystic pituitary mass. The pituitary macroadenoma extends upwards through the diaphragma, forming a waist, and distorts the optic chiasm in the suprasellar cistern. The infundibulum is distorted and the axoplasmic flow of pituitary hormones interrupted—note the posterior pituitary displacement into the suprasellar cistern (arrow, B). There is likely to be cavernous sinus invasion because one-third of cavernous internal carotid arteries will abut the tumour mass.
Approximately 10% compress or invade the cavernous sinus, resulting in cranial nerve palsies. Invasion can be difficult to judge, but encasement of >30% diameter of the internal carotid artery reliably indicates cavernous sinus invasion. Other indicators are tissue interposed between the cavernous internal carotid artery and the carotid sulcus, or if tissue crosses the line joining the medial cavernous and suprasellar internal carotid artery. Pituitary tumours extending into the cavernous sinus do not narrow the internal carotid artery, unlike meningiomas and inflammatory processes. Cavernous sinus invasion is important to recognise because surgery is then not curative and patients will require radiosurgery. Serum prolactin concentrations above 1000 µ/L (normal <360) also suggest cavernous sinus invasion.
They can also erode the dorsum and the central skull base, and if more extensive they invade the sphenoid sinus, clivus, nasal cavity, interpeduncular cistern, orbit and ethmoids.
Empty sella
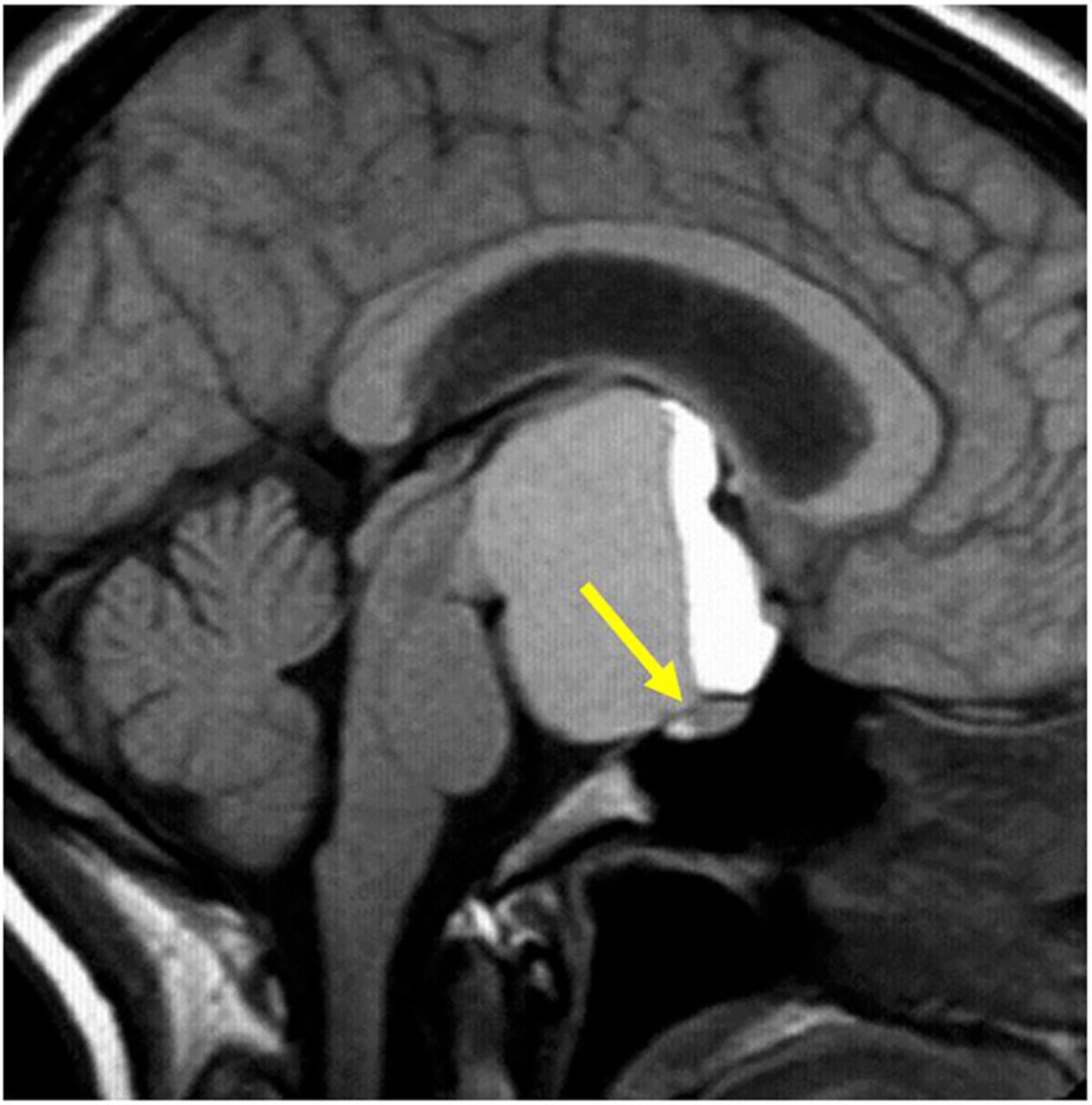
This is often a normal variant of little or no significance. Thinning or absence of the diaphragma allows the suprasellar cistern to herniate into the sella. Chronic CSF pulsation expands the fossa and flattens the gland (figure 4). The infundibulum is normal. Occasionally, the patient may show mild pituitary dysfunction.

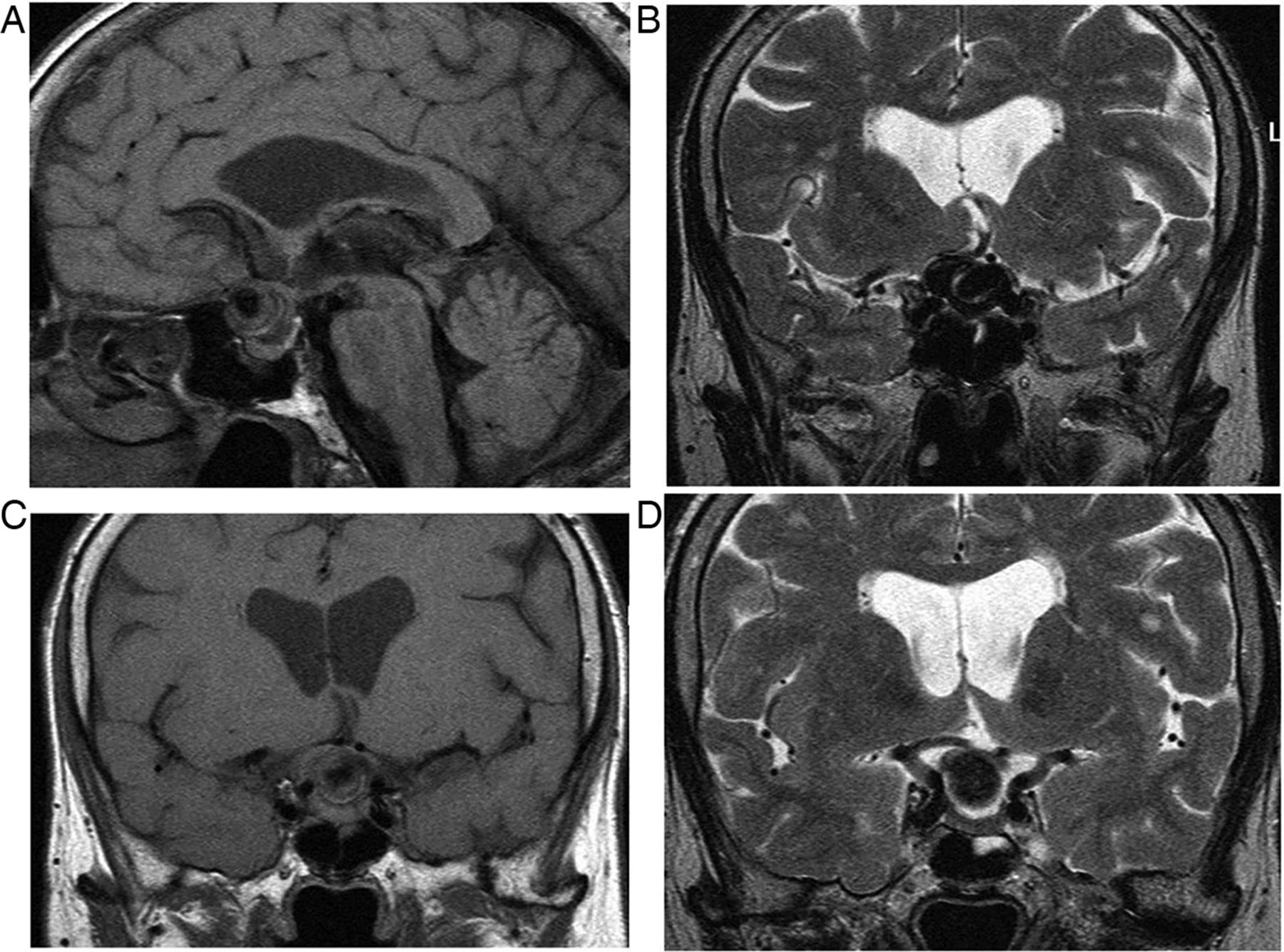
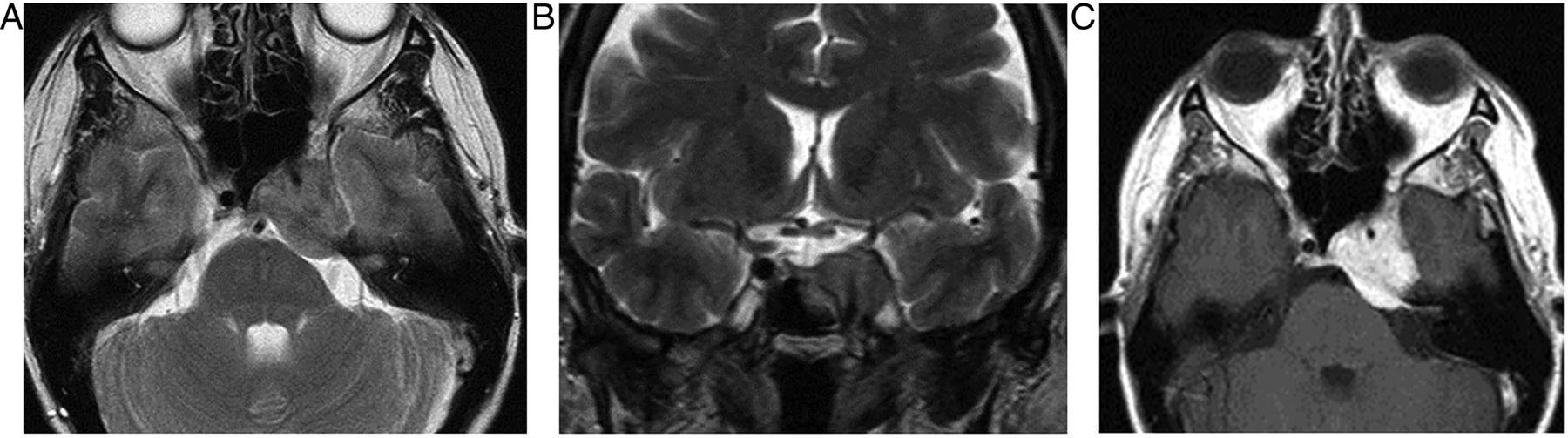
Primary partially empty sella. T1W sagittal (A), T1W (B) and T2W (C) coronal MRIs show expansion of the pituitary fossa and flattening of the gland along the sella floor. The infundibulum extends from the hypothalamus down to the flattened pituitary tissue, so distinguishing it from a cystic intrasellar mass.
A partially empty sella may develop in idiopathic intracranial hypertension. A secondary empty sella arises postoperatively, following radiotherapy and after macroadenoma infarction.
Rathke's cleft cysts
These are usually small cysts, found incidentally, and are usually stable. Rarely, they become large enough to compress surrounding structures (producing, as expected, headache, visual and endocrine disturbance). They are benign lesions arising from epithelial remnants of Rathke's pouch, which is a precursor of the anterior and intermediate lobes. They are well defined, spherical or ovoid, and are located centrally in the pars intermedia. They are either intrasellar (40%) or intrasellar and suprasellar; only a minority are purely suprasellar. The MR appearance is variable and depends upon cyst composition. They are lined by columnar or cuboidal epithelium, and their contents are either mucous or serous. Two-thirds are T1 hyperintense, reflecting the proteinaceous content (figure 2); one-third are T1 hypointense. Half are T2 hyperintense, 25% isointense and 25% T2 hypointense. A hypointense nodule within the hyperintense cyst is characteristic. Mild enhancement of the wall may result from inflammation or squamous metaplasia of cyst wall. They do not calcify.
Pituitary apoplexy
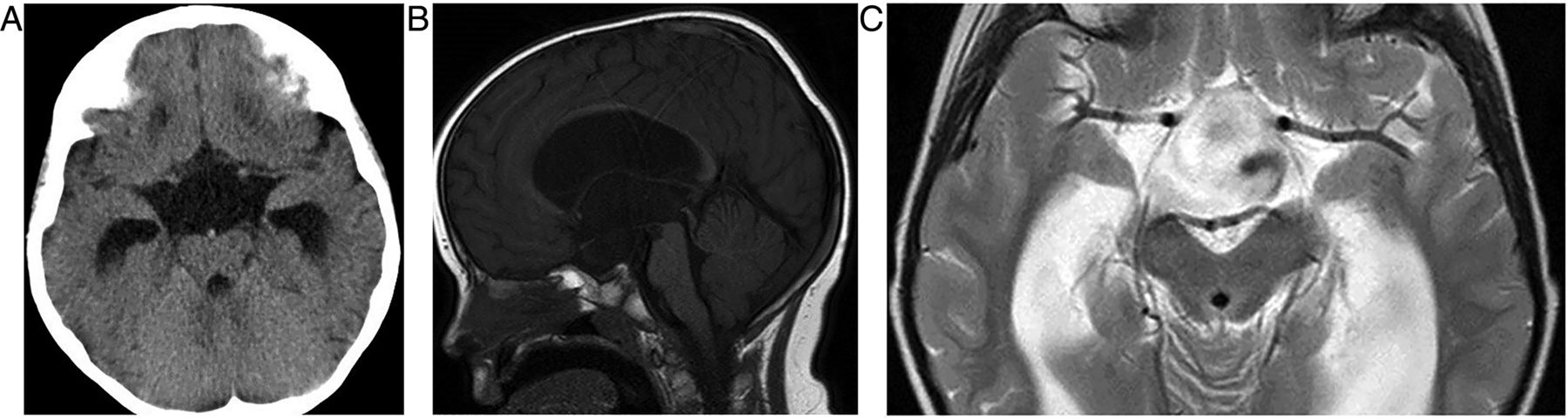
This a rare but potentially life-threatening event. Patients present with sudden onset of headache, visual changes, ophthalmoplegia and reduced consciousness; the condition can mimic subarachnoid haemorrhage, meningitis, stroke or migraine. The mean age at presentation is 49 years, and it affects mostly men. It results from acute infarction of the gland, which may be haemorrhagic. Usually the underlying cause is a pituitary adenoma that outgrows its blood supply. The result is electrolyte disturbance and a spectrum of endocrine disturbance.
Apoplexy complicates approximately 1% of macroadenomas; silent haemorrhage into an adenoma far more common.
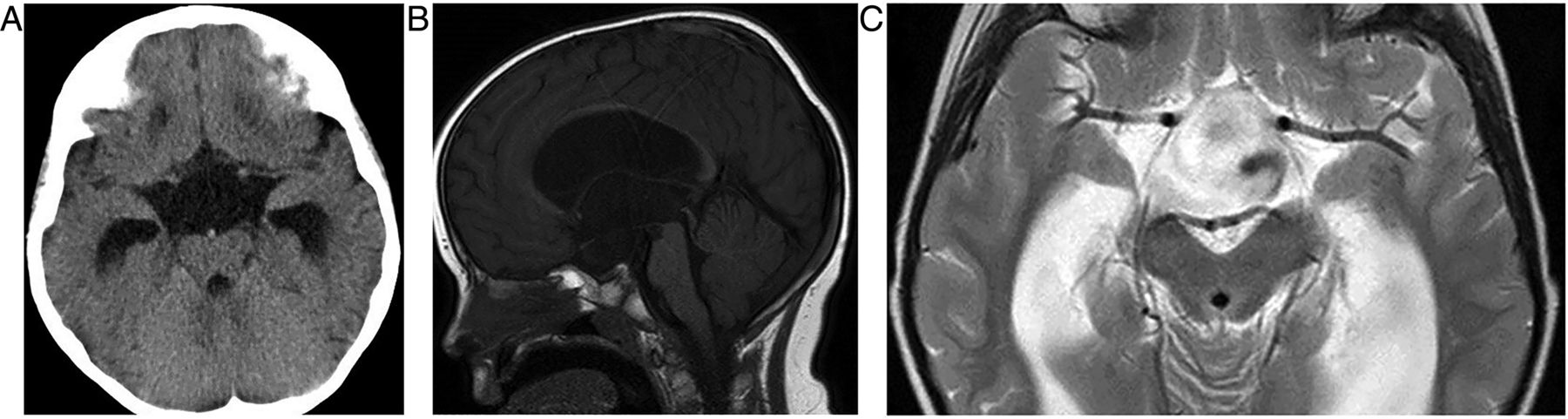
A CT scan shows a sellar mass and the sphenoid sinus may have thickened mucosa due to regional venous engorgement. MR scanning shows the mass and blood optimally (figure 5).

Pituitary apoplexy. T1W sagittal and T1W coronal images of the pituitary fossa show a pituitary mass, almost certainly a macroadenoma, which has expanded and remodelled the pituitary fossa and extended up into the suprasellar cistern where it compresses the chiasm. The macroadenoma has outgrown its blood supply and undergone haemorrhagic infarction. There is T1 hyperintense methaemoglobin diffusely within the macroadenoma. There is invasion of the right cavernous sinus (B, arrows). The patient presented with sudden-onset severe headache and visual loss.
Other risk factors for apoplexy include anticoagulants, radiotherapy, bromocriptine, trauma and the peripartum or postpartum period (Sheehan's syndrome). Postpartum apoplexy arises in a non-tumourous gland in women who have suffered significant postpartum haemorrhage with hypovolaemia. The pituitary gland normally hypertrophies in pregnancy and so is more susceptible to infarction from hypoperfusion.
Pituitary abscess
A pituitary abscess may arise secondary to haematogenous spread, directly or postoperatively from sphenoid sinusitis, cavernous sinus thrombosis, meningitis or occasionally in cystic adenomas. The most common organisms are Gram-positive cocci, but the abscess is sterile in half of the cases.
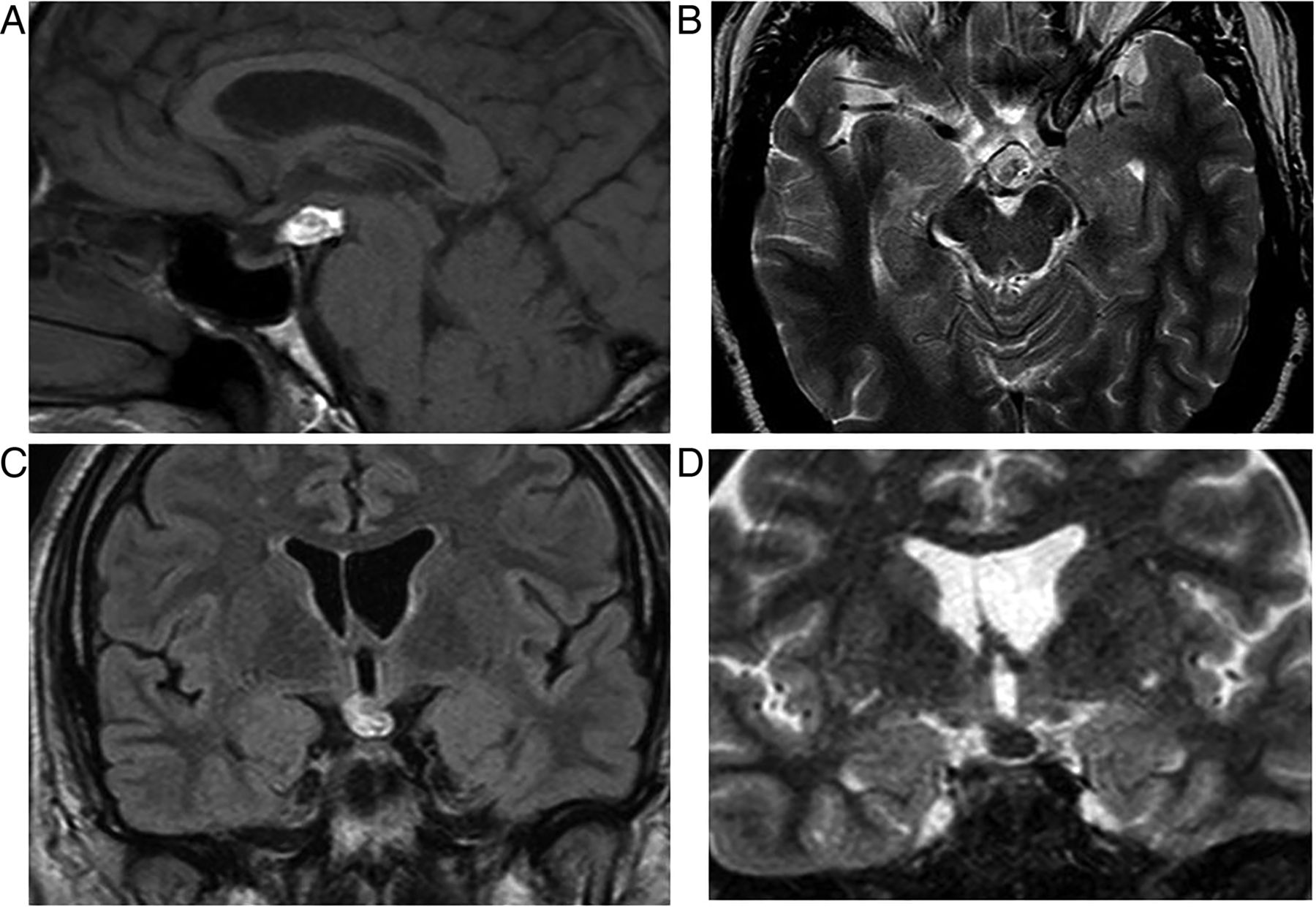
Lymphocytic hypophysitis
This is an idiopathic inflammatory, sometimes autoimmune, corticosteroid-responsive pituitary disorder, similar to orbital pseudotumour. It traditionally manifests in young, pregnant women who present with signs of a sellar mass, pituitary dysfunction, with signs and symptoms of headache, fever, nausea, vomiting, meningism, cranial neuropathy, visual disturbance and diabetes insipidus or hypopituitarism. CSF is lymphocytic.
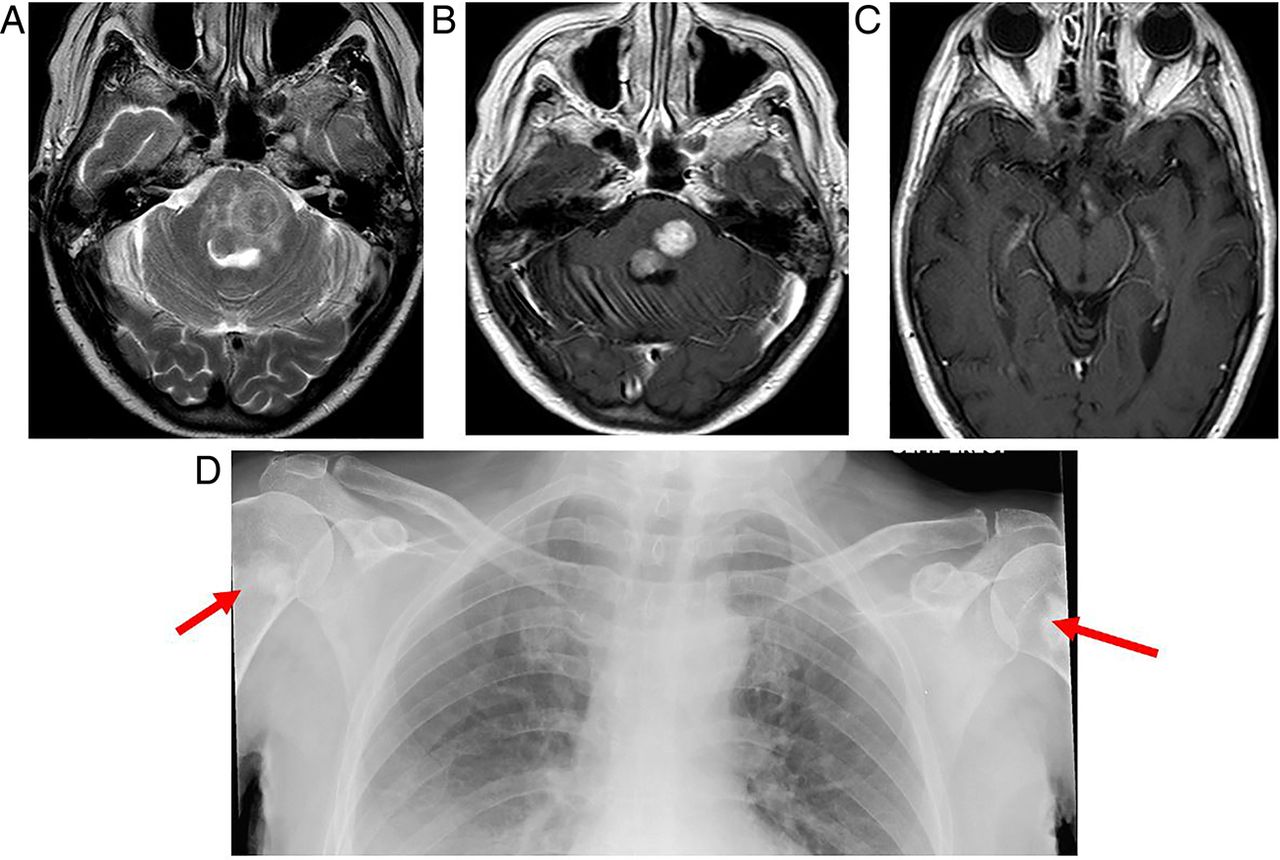
The pituitary gland is homogeneously enlarged by the inflammatory infiltrate (figure 6), comprising lymphocytes, monocytes and plasma cells. The stalk is infiltrated, and the bright spot of the posterior pituitary is absent. Often the enhancement is avid and homogeneous, extending along the infundibulum and hypothalamus and sometimes into the cavernous sinus. Occasionally cystic ring enhancement may even mimic an abscess or necrotic tumour.

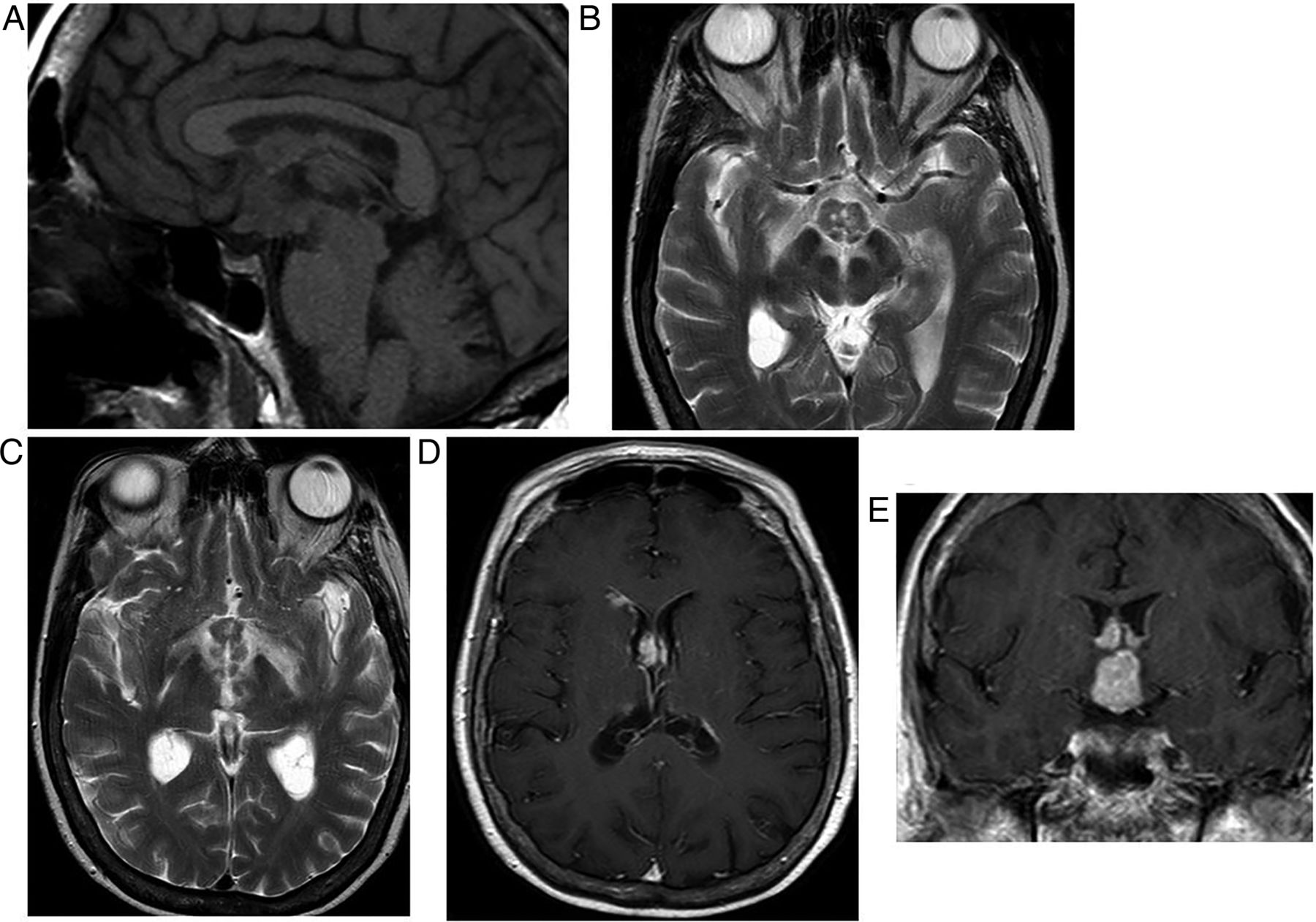
Lymphocytic hypophysitis. A 30-year-old woman presented with headache and visual loss and endocrine assessment confirmed panhypopituitarism. T1W sagittal (A) and coronal (B), T2W coronal (C and D) and gadolinium-enhanced T1W (E) MRIs show an enhancing bulky pituitary gland, absent posterior pituitary high signal (indicating central diabetes insipidus), thickening of the infundibulum and an enhancing optic chiasm/hypothalamic mass. There is oedema in the hypothalamus, which is tracking along the optic nerves. There is enhancement of the thickened dura as it passes posteriorly along the clivus. Six weeks later, after a course of corticosteroids, there is a dramatic improvement with only a small amount of residual enhancing tissue (F, T1W gadolinium-enhanced sagittal MRI).
Hypothalamic and other suprasellar disorders
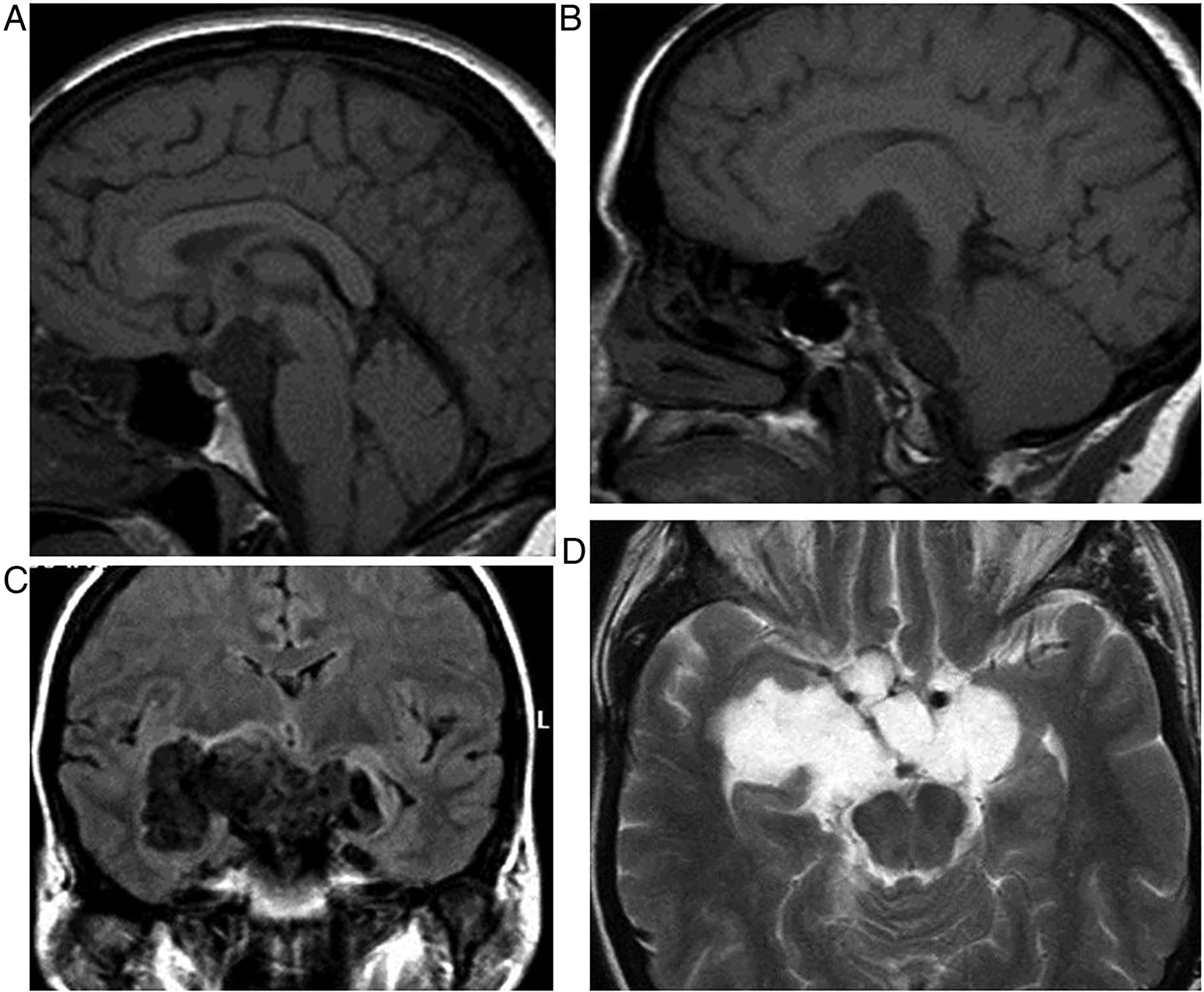
Craniopharyngiomas
These are the most common suprasellar mass, and although usually entirely suprasellar, one-third may extend into the sella; 10% are confined to the sella. They are benign, slow-growing, epithelial tumours arising from the remnants of Rathke's pouch. Their presentation relates to both their size and their precise location, and includes headache, a visual field defect, visual loss and endocrinopathy.
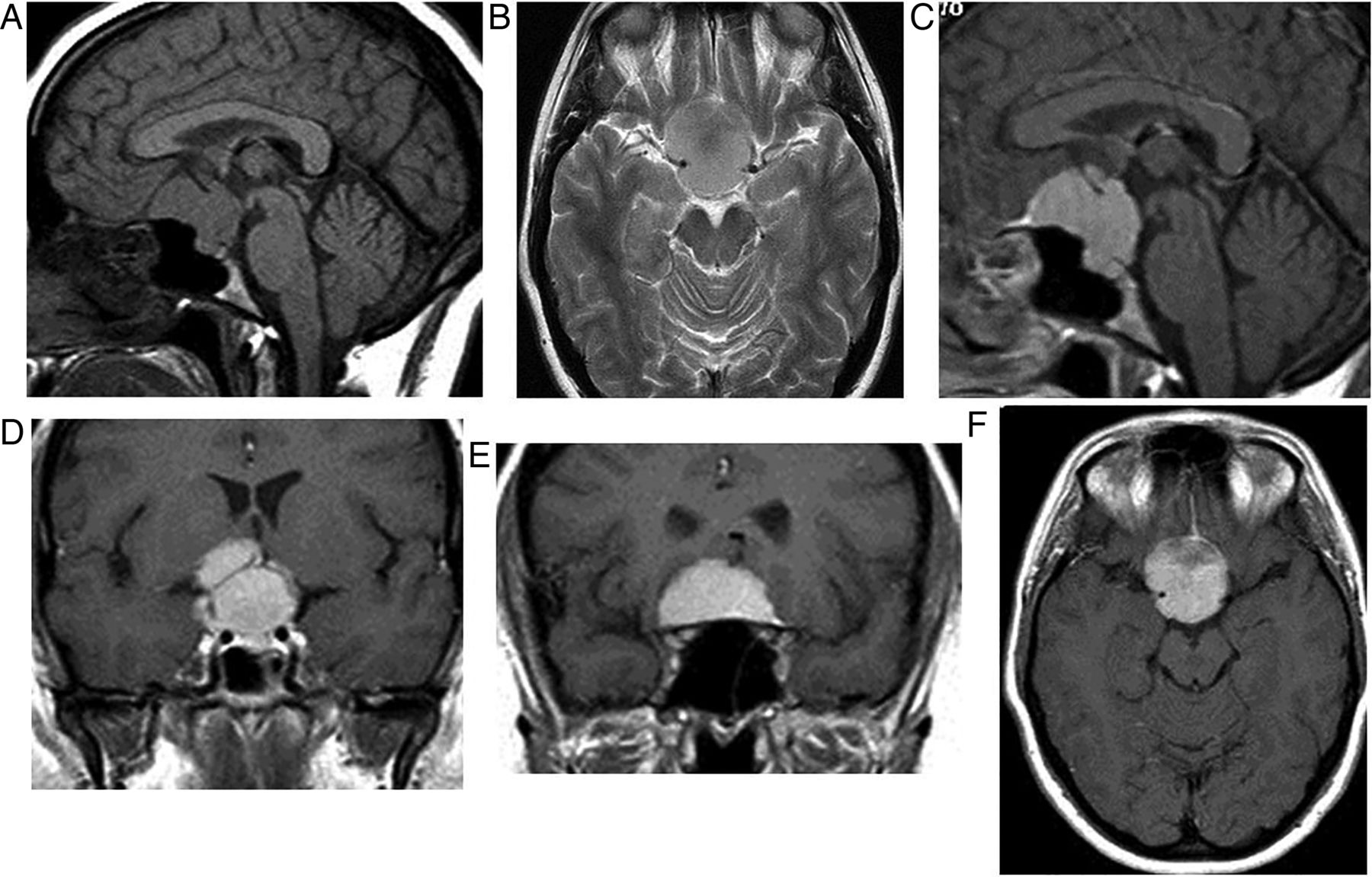
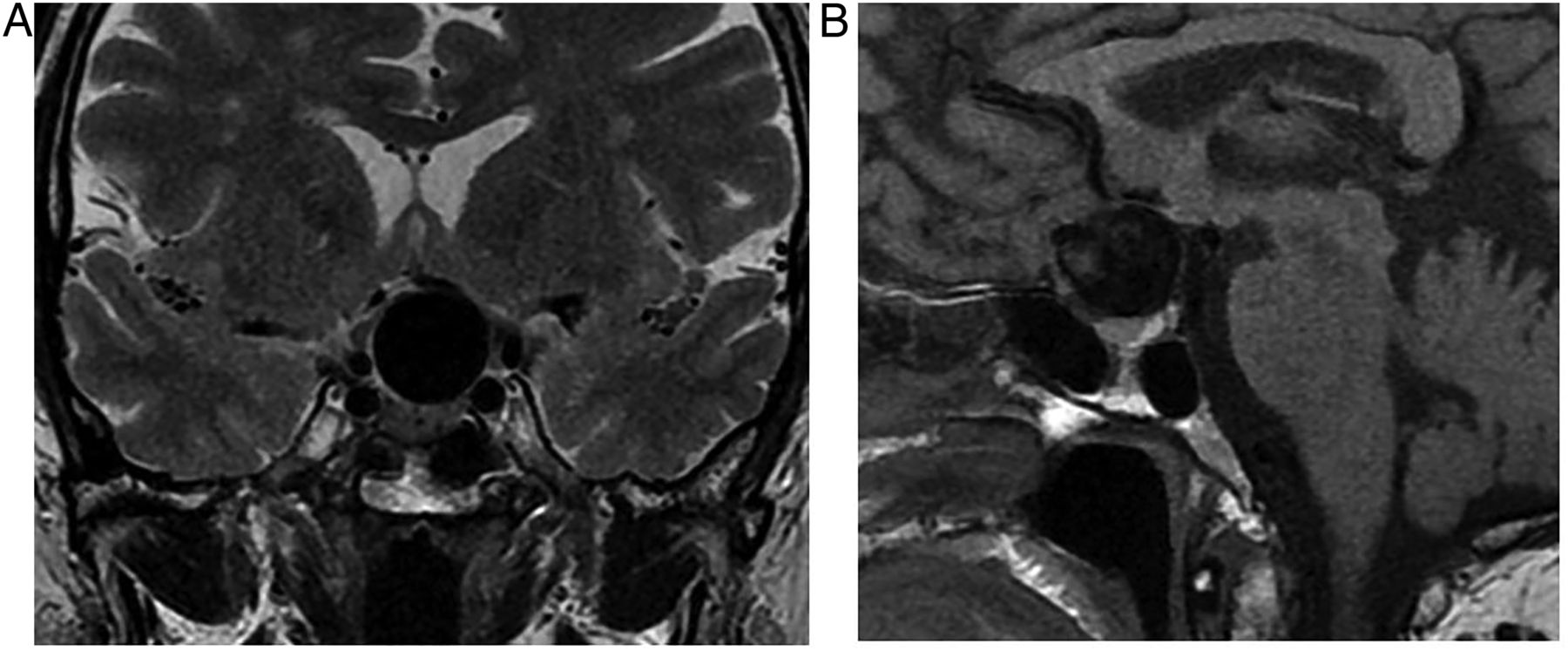
There is a bimodal age distribution: 5–15 and 65–74 years of age. They are completely or partly cystic. The well-demarcated, often loculated cysts may contain cholesterol, may be haemorrhagic or can be highly proteinaceous, and so they often show high signal on T1W (figure 7). Ninety per cent calcify.
Craniopharyngioma: T1W sagittal MRI shows a large complex suprasellar cystic mass with a fluid level. Note the T1 hypointense diaphragma separates the pituitary gland from a suprasellar mass. The pituitary fossa is normal in size. There is a high signal posterior pituitary. The mass compresses the optic chiasm and distorts the hypothalamus. It is likely that the very high T1 signal material is cholesterol floating on proteinaceous fluid.
The adamatinous type occurs mostly in children. These are usually predominantly cystic; oedema may develop along the optic tract.
The squamopapillary type occurs mostly in adults. They comprise predominantly solid, enhancing tissue with small cysts.
Meningiomas
Meningiomas are the second most common parasellar mass, arising from a variety of sites: the tuberculum sella, floor of the anterior cranial fossa, sphenoid wing, cavernous sinus (where they may encase and narrow the internal carotid artery), dorsum sella and clinoid processes.
They are usually homogenous, T1 isointense or hypointense compared with grey matter and T2 isointense or hyperintense compared with grey matter. They avidly enhance with gadolinium and have a broad-based dural attachment (figure 8). Some are calcified, some are cystic and some generate bone hyperostosis or lysis.
Planum meningioma. T1W sagittal (A), T2W axial (B) and gadolinium-enhanced T1W sagittal (C), coronal (D and E) and axial (F) MRIs show a typical planum meningioma—a broad-based extra-axial mass, returning similar signal to cortex, uniformally brightly enhancing, with a dural tail anteriorly (C, arrow). Meningioma extends down into the pituitary fossa and into the suprasellar cistern, compressing the optic nerves and chiasm and displacing both anterior cerebral arteries superiorly. The T1-hypointense diaphragma clearly separates the suprasellar mass from the pituitary gland. The fossa size is normal. These factors confirm the origin is suprasellar rather than intrasellar.
Cavernous sinus lesions can extend into the pontine cistern via the porous trigeminus. Reactive enhancing dural tails can occur with pituitary macroadenomas, and so are not pathognomonic for meningiomas.
Arachnoid cysts
Fifteen per cent of arachnoid cysts are suprasellar. They arise by splitting the arachnoid membrane and so contain CSF. They can progressively enlarge, producing visual, endocrine disturbance and hydrocephalus, obstructing the foramen of Munro (figure 9).
Suprasellar arachnoid cyst. Axial cranial CT scan (A), T1W sagittal (B) and T2W axial (C) MRIs show a thin-walled suprasellar cystic mass, containing cerebrospinal fluid only. There is pulsation artefact within the cyst. The cyst has compressed and thinned the hypothalamus (even the mamillary bodies are not visible) and it is obstructing the foramen of Munro with resultant hydrocephalus.
Lipomas
These benign fatty masses are derived from remnants or maldevelopment of the primitive meninx. In the parasellar region, they adhere to the infundibulum, floor of the third ventricle or adjacent cranial nerves. They are usually incidental and become symptomatic only when large (figure 10).
Lipoma. T1W sagittal (A), T2W axial (B), fast fluid-attenuated inversion recovery (FLAIR) coronal (C) and short tau inversion recovery (STIR) coronal (D) MRIs show a T1 high-signal hypothalamic mass. It returns the same signal as fat on all sequences and its signal is nulled (appears dark) on STIR, confirming its fatty nature.
They are well-defined, fatty-density lesions on CT and may show rim calcification. There is no enhancement. They are homogeneous with high T1 signal on MR. The fatty content is confirmed by chemical shift artefact and fat suppression techniques, which null the signal. This distinguishes them from haemorrhagic or proteinaceous lesions, which are also high signal on T1W.
Epidermoid cysts
These developmental epithelial inclusion cysts contain desquamated epithelial lining, cellular debris, keratin and cholesterol. The parasellar location is the second most common after the cerebellopontine angle. Patients present in their 30s and 40s with visual problems, hypopituitarism, diabetes insipidus and cranial nerves palsies. The imaging characteristics are identical to those lesions seen in the cerebellopontine angle (figure 11).
Epidermoid. T1W sagittal (A and B), fast fluid-attenuated inversion recovery (FLAIR) coronal (C) and T2W axial (D) MRIs show a parasellar epidermoid. These insinuating frond-like lesions are isointense or slightly hyperintense to cerebrospinal fluid (CSF) on T1W and similar to CSF on T2W, hyperintense to CSF on FLAIR and show restricted diffusion (not shown here). The hypothalamus is displaced upwards and the chiasm distorted. Note the invagination into the choroidal fissures bilaterally and extension inferiorly into the pontine cistern.
Dermoids
These are mostly midline, benign developmental, congenital inclusion cysts lined by stratified squamous epithelium and containing dermal elements—hair, sweat, sebaceous glands, fat and calcifications. They do not usually enhance. They present earlier than epidermoids, usually in young men (figure 12) in their teens, with optic neuropathy, visual field defect, diabetes insipidus and/or hypopituitarism.
Dermoid. T1W sagittal (A), coronal fast fluid-attenuated inversion recovery (B) and T2W axial (C) MRIs show a complex extra-axial mass arising from the region of the anterior clinoid and planum on the right and extending into the anterior middle cranial fossa. It is predominantly high T1 signal, reflecting its fatty content. There is some lower signal, possibly from hair. It abuts the right optic nerve, chiasm and tract. Like epidermoids, these occasionally may rupture into the subarachnoid space, resulting in a chemical meningitis.
On imaging, they are well-circumscribed, usually heterogenous cystic lesions containing fat and calcifications.
Parasellar aneurysms
There are numerous arteries in the parasellar region. Arterial aneurysms may arise from the cavernous internal carotid artery, basilar, posterior cerebral, posterior communicating, anterior communicating and ophthalmic arteries (figure 13). The latter two may cause visual loss and/or a visual field defect. Giant cavernous aneurysms may cause a painful ophthalmoplegia.
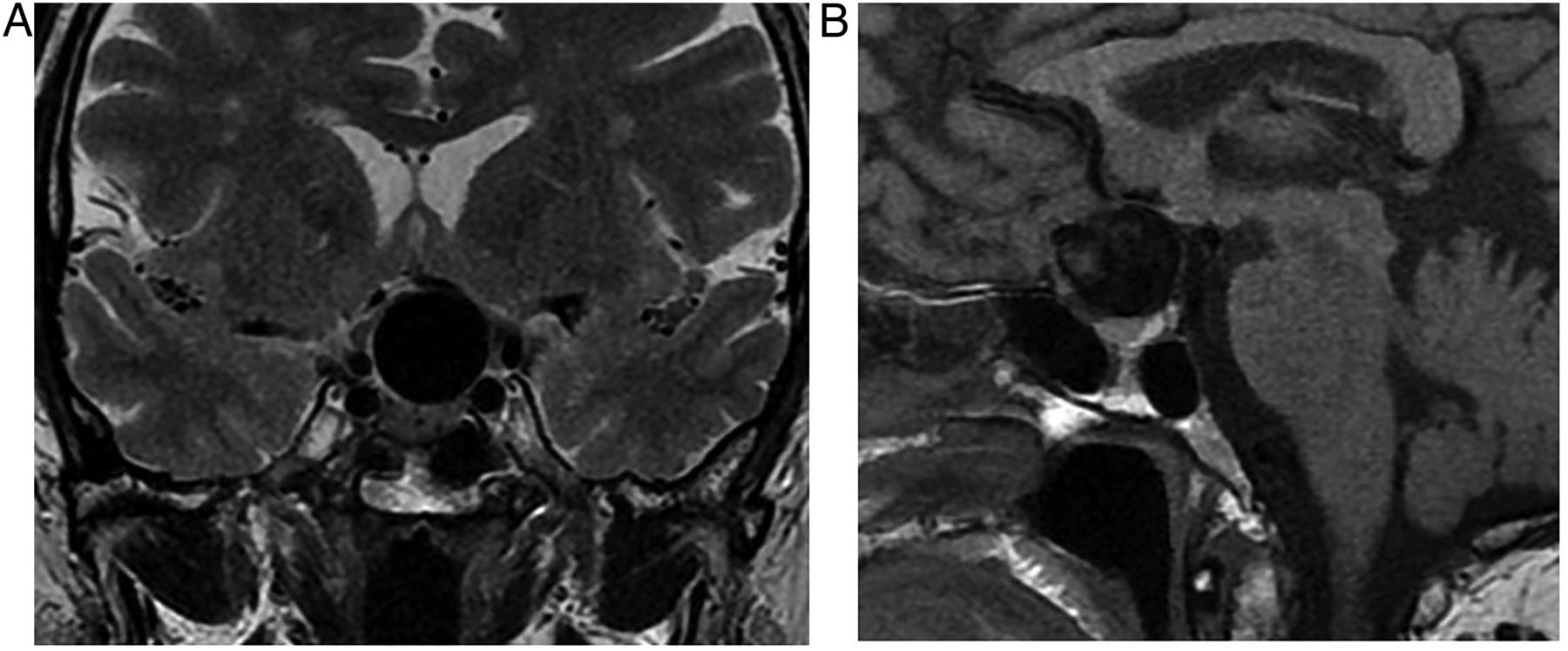
Giant ophthalmic aneurysm. T1W coronal (A) and sagittal (B) MRIs in a 40-year-old woman presenting with progressive left eye visual loss due to a giant left ophthalmic aneurysm. There is a large signal void—the aneurysm sac—without thrombus, in the suprasellar cistern. The chiasm and left optic nerve are compressed.
A classical pupil-involving painful third nerve palsy may result from its compression by a posterior communicating or basilar termination region aneurysm.
MR imaging shows a flow void and phase artefact, sometimes with laminated thrombus (figure 14) containing foci of T1 hyperintensity, T2 hypointensity or hyperintensity, reflecting blood products (methaemoglobin and haemosiderin).
Large, partly thrombosed suprasellar ophthalmic aneurysm in a patient with visual failure. T1W sagittal (A) and coronal (C) and T2W coronal (B and D) MRIs show a suprasellar mass with a flow void, compressing the optic chiasm. Laminated thrombus gives rise to complex signal intensities reflecting methaemoglobin, haemosiderin and calcification.
On CT scanning, they appear as round or ovoid, lobulated, mildly hyperdense, enhancing lesions, with or without calcification.
Hypothalamic gliomas
These are usually slow-growing, low-grade diffuse tumours of childhood (figure 15), presenting with poor visual acuity, endocrine dysfunction or a diencephalic syndrome. Half have neurofibromatosis type 1 (figure 16). They are usually large when first diagnosed. Most are juvenile pilocystic astrocytomas. They are usually T1-hypointense, T2-hyperintense and show varying degrees of enhancement and cystic change.
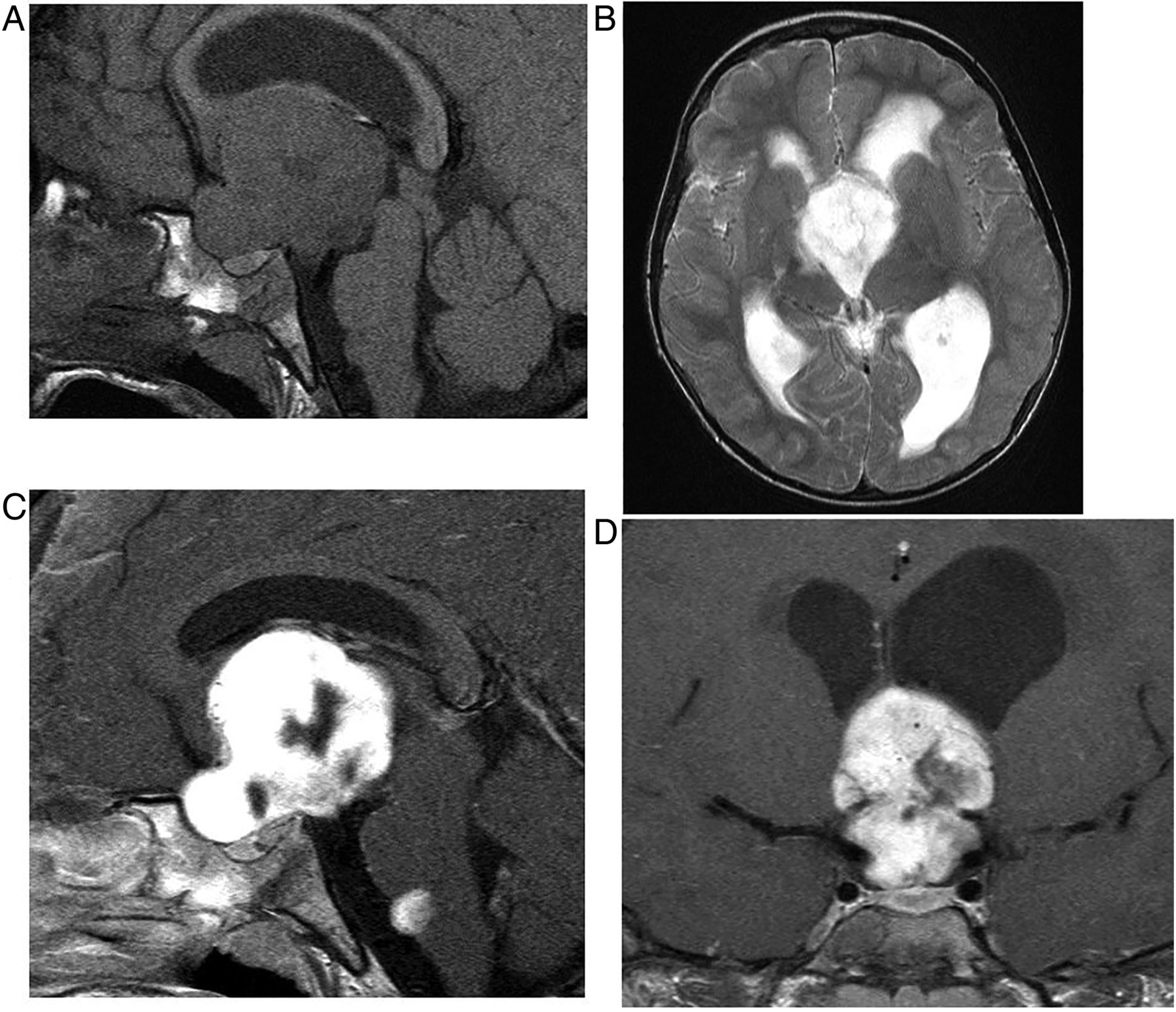
Hypothalamic glioma (pilocystic astrocytoma). T1W sagittal pre (A) and post-gadolinium (C), T2W axial (B) and T1W gadolinium-enhanced coronal (D) MRIs show a large, brightly but inhomogeneously enhancing hypothalamic mass with a drop metastasis at the pontomedullary junction. The mass obstructs the foramen of Munro, causing hydrocephalus.

Hypothalamic/optic pathway glioma in a patient with neurofibromatosis type 1. T1W sagittal (A), T2W coronal (B and C) and T2W axial MRIs show a solid mass arising from the hypothalamus and extending anteriorly to involve the optic chiasm and right optic nerve (C, arrow). Note the normal-appearing left optic nerve (B, arrow). Note also the T1-hypontense diaphragma separating the anterior pituitary from the suprasellar mass. The T2-hyperintense foci, typical of dysmyelination in the basal ganglia and thalami, are pathognomonic of neurofibromatosis type 1 (D).
Germinomas
These are derived from primordial germ cell rests. They present most frequently around 10–12 years of age but may develop in young adults and can present with visual and endocrine disorders. They are midline lesions and 80% are pineal. The hypothalamus is the second most common site of origin, and they have no sex predominance (unlike their pineal counterparts). They are synchronous in 10% (figure 17).

Synchronous hypothalamic and pineal germinomas. T1W MRI shows T1 isointense pineal and hypothalamic masses. The hypothalamic mass extends to involve the infundibulum and pituitary gland. The posterior pituitary high signal is absent—the patient had central diabetes insipidus. The pineal mass compressed the aqueduct and posterior third ventricle, without hydrocephalus. Gadolinium-enhanced images of the brain and spine showed avid enhancement of both lesions but no leptomeningeal dissemination.
On MR, they are infiltrating lesions. On T1W imaging, they return a signal similar to cortex and on T2W they are slightly hyperintense to cortex. They enhance avidly and sometimes heterogeneously. Cysts, calcification and haemorrhage are rare. One-third show restricted diffusion. They may disseminate throughout the subarachnoid space.
Sarcoidosis
Sarcoidosis involves the central nervous system in 5–15% of patients. It is an infiltrative process with non-caseating granulomas predominantly affecting the leptomeninges, creating a granulomatous basal meningitis (figure 18). This classically involves the hypothalamus, infundibulum and optic chiasm, causing diabetes insipidus and visual problems. In some patients, there are dural-based masses, thickened cranial nerves and periventricular multifocal white matter lesions.
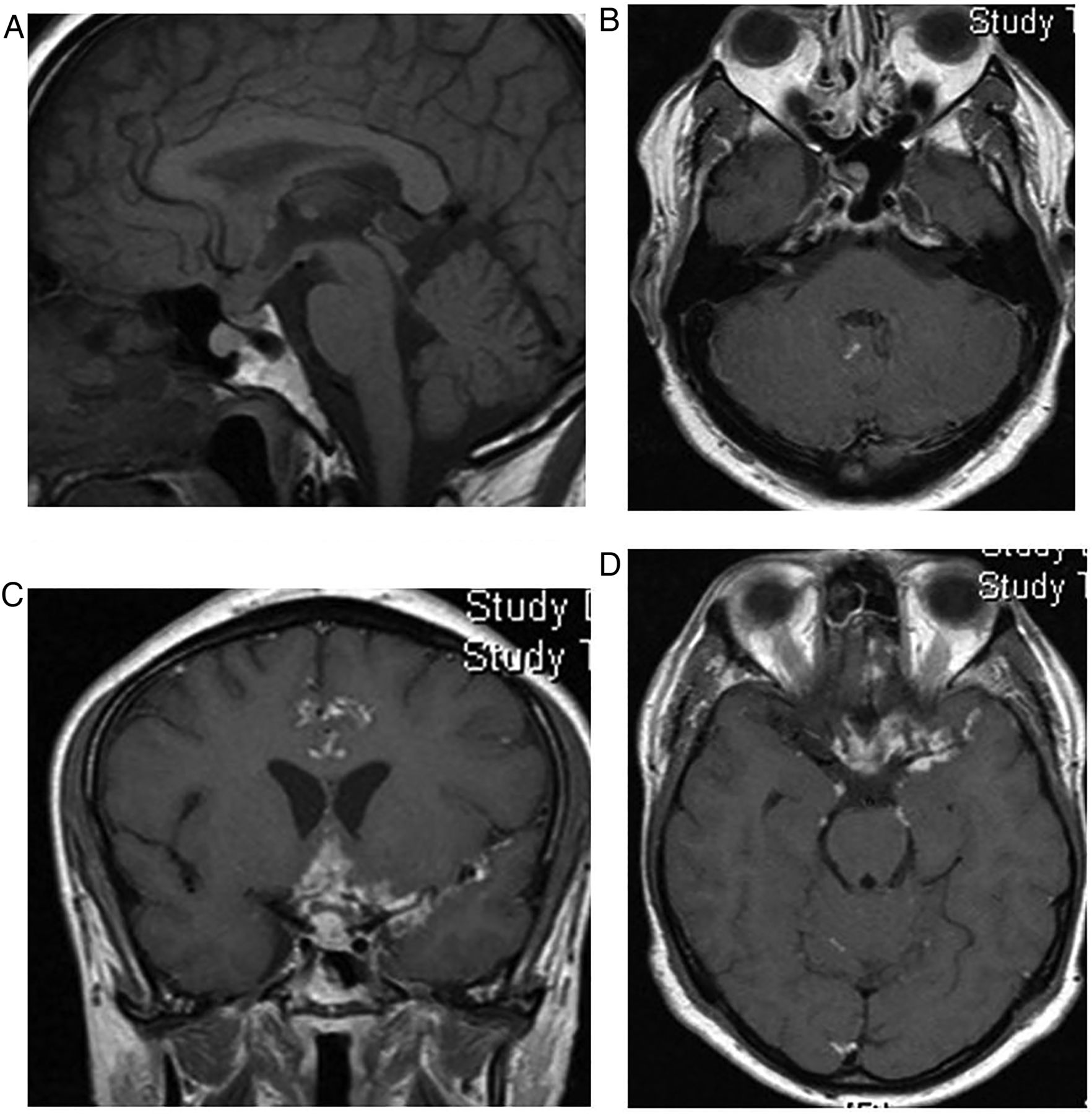
Sarcoidosis. T1W sagittal and gadolinium-enhanced T1W axial (B and D) and coronal (C) MRIs show an infundibular mass and nodular enhancement of the basal meninges, suggesting a granulomatous basal meningitic process. The high signal of the posterior pituitary is absent, confirming central diabetes insipidus. Note an enhancing lesion in the right internal auditory meatus.
Other granulomatous disorders that may involve the hypothalamus and basal meninges include tuberculosis, histiocytosis, Wegener's granulomatosis (granulomatosis with polyangiitis), Rosai–Dorfman and Erdheim–Chester disease (figure 19).

Erdheim–Chester disease. This is a rare systemic non-Langerhans histiocytosis of unknown aetiology, involving multiple organs, with a predilection for bones, orbits and brain. In the brain, there may be infiltrative lesions and/or mass lesions affecting mainly the dentate and parasellar regions or meningeal-based masses with either thickening of the dura or meningioma-like tumours. If the diagnosis is not already known, the diagnosis of the extra-axial masses is difficult. The combination of dural lesions, intraorbital and parasellar masses and osteosclerotic changes of the bones in a patient with a cerebellar syndrome might suggest the diagnosis. T2W axial (A) and T1W axial gadolinium-enhanced (B and C) images show low T2 signal, enhancing masses in the pons, associated with oedema and nodular enhancement of the hypothalamus (B). Sclerotic lesions in the humeral necks (arrows, D) suggested the diagnosis.
Malignant meningitis may show imaging changes similar to sarcoidosis (figure 20).
Malignant meningitis from lymphoma. T1W sagittal, T2W axial (B and C) and T1W gadolinium-enhanced axial (D) and coronal MRIs (E) show an enhancing chiasm/hypothalamic mass associated with oedema and enhancing masses in the frontal horns of the lateral ventricles. The patient-reported rapid deterioration in vision, cognitive problems and central diabetes insipidus. Rapid progression favours a neoplastic process. Breast and lung are the most common primaries to produce malignant meningitis.
Hypothalamic hamartoma
This is a pedunculated or sessile, developmental, non-neoplastic lesion (prevalence 1–2 per 100 000) comprising grey matter with both small and large neurones, myelinated and unmyelinated nerve fibres and glial tissue, located between the infundibulum and mamillary body, in the tuber cinereum (figure 21). It can occasionally project upwards into the third ventricle and may grow slowly. The small spontaneously-firing GABA-ergic neurones contribute to its epileptogenicity. Impulses propagate to the hippocampal formations or via the mammillothalamic tract to the anterior thalamus and the cingulate gyrus.

Hamartoma of the tuber cinereum. T1W sagittal MRI in a young person with seizures confirms the aetiology. A pedunculated mass arises from the tuber cinereum. It is homogenous, returns similar signal to cortex and does not enhance.
It returns signal similar to grey matter on T1W MR but is slightly T2W and fast fluid-attenuated inversion recovery (FLAIR) hyperintense. There is no enhancement. Cystic change and calcification is rare.
As a consequence of its location, it may present not only with seizures but also with endocrine disturbance, precocious puberty (some contain luteinising hormone-releasing hormone granules), behavioural, cognitive and psychiatric disorders.
Cavernous sinuses
Cavernous sinuses comprise two layers of dura, which split to form a venous channel, extending from the orbital apex and superior orbital fissure to Meckel's cave, lateral to the pituitary fossa. They are multilocular, compartmentalised, venous structures receiving blood from the ophthalmic veins, middle cerebral veins and sphenoparietal sinus and draining to the petrosal sinuses and pterygoid plexus. There are intracavernous connections. The cavernous sinuses contain the third, fourth, sixth and the first and second divisions of the fifth cranial nerves and occasionally fat, together with the cavernous internal carotid artery. The Gasserian ganglion is located posteriorly in Meckel's cave, a dura reflection protruding into the cavernous sinus.
Carotico-cavernous fistulas
These are abnormal connection between the carotid artery and cavernous sinus. They may be high flow and direct—secondary to trauma or rupture of a cavernous aneurysm—or low flow, dural in type and involving branches of the external carotid artery and/or dural branches of the internal carotid artery (figure 22).

Left-sided dural carotico-cavernous fistula in a patient with tinnitus and a left sixth nerve palsy. T1W gadolinium-enhanced axial images, with fat saturation, show numerous small vascular flow voids—multiple small dural arterial feeders, in the lateral wall of the cavernous sinus. Venous drainage is posterior via a dilated inferior petrosal sinus (arrow).
As a result of arteriovenous shunting and venous congestion, patients develop tinnitus and signs of orbital congestion, with a pulsating exophthalmoplegia and chemosis. If the intraocular pressure becomes very high, the visual acuity deteriorates, sometimes with visual loss. Retrograde filling of the middle cerebral veins risks intracranial haemorrhage.
Tolosa–Hunt syndrome
This is a retro-orbital pseudotumour, a low-grade inflammatory, granulomatous, corticosteroid-responsive process involving the cavernous sinus and orbital apex. The process is usually unilateral (but can be bilateral) and causes severe painful ophthalmoplegia and cranial nerve palsies.
The enlarged cavernous sinus contains soft tissue granulomatous masses (lymphocytes and plasma cells), is isointense to muscle on T1W, shows variable signal on T2W and enhances with gadolinium (figure 23). A secondary arteritis may narrow the cavernous internal carotid artery, with thickening of its arterial wall.

Tolosa–Hunt in a patient with left-sided, painful ophthalmoplegia. Gadolinium-enhanced T1W axial (A) and coronal (B–D) MRIs, with fat saturation, show increased enhancement in the left cavernous sinus and orbital apex. The internal carotid artery is not narrowed.
Schwannomas
These benign tumours arise from Schwann cells, usually from the trigeminal nerve. They present with facial numbness, pain and paraesthesia.
The schwannoma may be solid or necrotic/cystic, occasionally haemorrhagic and is avidly enhancing (figure 24). Before contrast, it returns signal similar to grey matter on T1W and is relatively hyperintense on T2W. They are fusiform and well marginated, but when in Meckel's cave they can be dumbbell in shape and extending into the pontine cistern with a waist at the porous trigeminus. They follow the expected course of the nerve. They do not narrow the cavernous internal carotid artery, unlike cavernous sinus meningiomas (figure 25).

Trigeminal schwannoma. T2W axial (A) and T1W axial gadolinium-enhanced axial MRIs show a cystic, brightly enhancing, right cavernous sinus mass, extending posteriorly along the course of the trigeminal nerve.
Left cavernous sinus meningioma. T2W axial (A) and coronal (B) and T1W gadolinium-enhanced MRIs show an enhancing cavernous sinus mass constricting the left cavernous internal carotid artery, typical of a meningioma. Enhancing tissue extends into the pontine cistern.
Cavernous haemangiomas
Cavernous haemangiomas are most common in women in their 40s. The tumour, attached to the outer wall of the cavernous sinus, is formed by endothelial-lined sinusoidal spaces, containing slow flowing or stagnant blood. They are hyperintense on T1 and T2—unlike meningiomas and schwannomas—usually with progressive ‘filling in’ after contrast or may show homogeneous or heterogeneous contrast enhancement.
The skull base
The skull base, being a thin bony structure, is usually best imaged using a combination of CT (using bone windows) and MR. The best MR sequences are T1W axial gadolinium-enhanced images with fat saturation and short tau inversion recovery (STIR), or T2W images with fat saturation, both in the axial and coronal planes.
Clival neoplasms
Clival neoplasms may involve the parasellar region and include chordoma, metastasis, myeloma, chondrosarcoma, lymphoma and meningioma.
Chondrosarcoma
Chondrosarcoma is a slow-growing neoplasm that is less common than chordoma. Patients may present with ophthalmoplegia. They are para-midline, malignant tumours arising from the cartilage-forming cells of the spheno-occipital synchondrosis (figure 26) and petroclival synchondrosis; they invade the sphenoid and destroy the clivus. Chondrosarcomas comprise 6% of skull base tumours and 75% are parasellar. They are isointense to hypointense on T1W with high T1W foci reflecting mucin and haemorrhage. They are T2 hyperintense with low T2 foci corresponding to the stippled calcified chondroid matrix. They enhance markedly and heterogeneously usually.

Chondrosarcoma: T2W axial (A and B) and fast fluid-attenuated inversion recovery coronal images show a large T2 hyperintense left cavernous sinus mass, extending upwards from the left petroclivical synchondrosis. The low T2 foci correspond to the stippled calcified chondroid matrix. The Eustachian tube is blocked with secondary inflammatory changes in the mastoid air cells. Note the atrophy of the masseter, temporalis and pterygoids, reflecting the left trigeminal nerve dysfunction. There was also left tongue denervation (not shown) due to involvement of the left hypoglossal canal/nerve. There is oedema in the left temporal lobe.
As with most bony skull base pathologies, CT confirms their site of origin, shows bony destruction and curvilinear chondroid calcification in the soft tissue matrix.
Chordomas
Chordomas are rare, slow-growing tumours arising from primitive notochord elements in the clivus near the spheno-occipital synchondrosis. The most common presenting symptom is diplopia, due to a sixth nerve palsy, because the tumour compromises Dorello's canal. CT scan of head shows extensive bone destruction, a soft tissue mass and tumoural calcification. MR scanning shows a mass of moderate T1 and high T2 signal with variable enhancement (figure 27). This locally invasive mass, which can extend into the cavernous sinus, may contain residual bony debris or sequestra, which return low T2 signal. There may also be haemorrhagic foci and highly proteinaceous cystic areas (high on T1W, high or low on T2W).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Chordoma. T1W axial (A) and coronal (B), T2W axial (C) and coronal (D) and gadolinium-enhanced T1W axial (D) MRIs show a mass of moderate T1 and high T2 signal with enhancement. This locally invasive mass destroys the clivus and sphenoid sinus and extends into the nasopharynx, pituitary fossa and cavernous sinuses. There are haemorrhagic foci and highly proteinaceous cystic areas (high on T1W).
Conclusion
Patients with parasellar disease may present to neurology because of visual loss, visual field defects, ophthalmoplegia, headache and seizures, resulting from compression of parasellar structures by a parasellar mass. Although many neoplastic, vascular and inflammatory processes may originate from parasellar structures, pituitary adenomas, craniopharyngiomas and meningiomas are the most common.
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; internally peer reviewed.
Linked Articles
- Editors' commentary
Other content recommended for you
- Neurology of the pituitary gland
- A case of symptomatic Rathke's cyst
- Spontaneous and asymptomatic rupture of an RCC with resolution of symptoms
- Spontaneous subarachnoid hemorrhage due to ruptured cavernous internal carotid artery aneurysm after medical prolactinoma treatment
- Granulomatous hypophysitis: a case of severe headache
- Pituitary infundibular epidermoid cyst: a rare cause of hypopituitarism
- Pituitary abscess
- Disorders of the anterior visual pathways
- An unusual cure for acromegaly
- Infundibulohypophysitis in a man presenting with diabetes insipidus and cavernous sinus involvement