Article Text

Abstract
Contractures are one of a handful of signs in muscle disease, besides weakness and its distribution, whose presence can help guide us diagnostically, a welcome star on the horizon. Contractures are associated with several myopathies, some with important cardiac manifestations, and consequently are important to recognise; their presence may also provide us with a potential satisfying ‘penny dropping’ diagnostic moment.
- myopathy contracture
Statistics from Altmetric.com
Patient 1
A 23-year-old man, who has never been able to run more than a few yards, has often fallen and struggled to jump as a child, arrives at clinic stating that recently climbing stairs has become more difficult and he can no longer help with heavy holiday luggage. His father had been a wheelchair user and needed a nocturnal ventilator after about 60 years of age.
On examination, he has thin muscles, no myotonia and mild proximal limb and neck flexion weakness; reflexes are reduced and sensation is normal.
Nerve conduction is normal, but his electromyography (EMG) looked myopathic; serum creatine kinase (CK) is at the upper limit of normal. A muscle biopsy shows a mild myopathy but leads to no clear diagnosis.
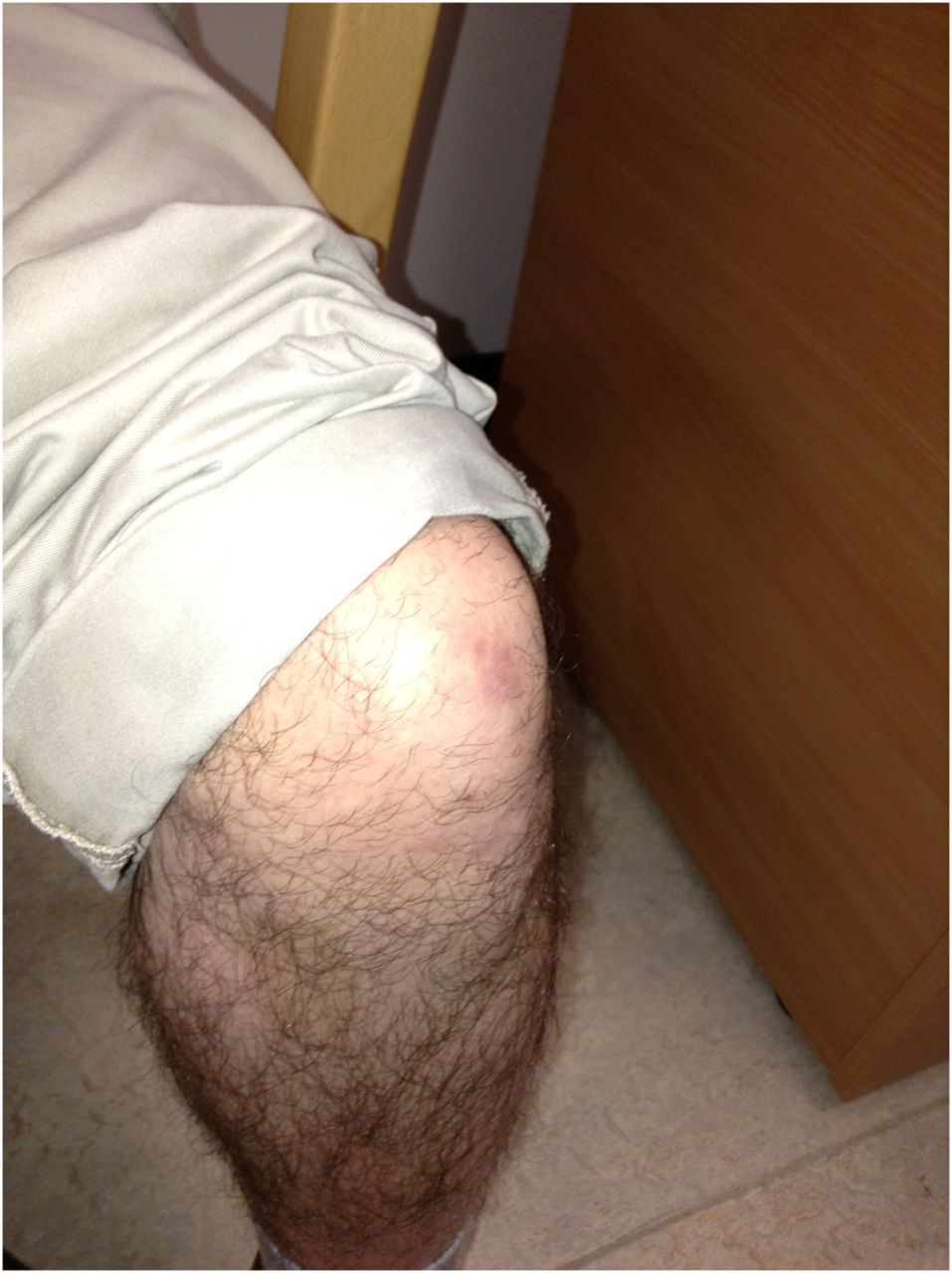
At this point, in the best neurological tradition, you revisit the history and examination and find contractures (see figures 1⇓⇓–4), a denouement that sets you on the right diagnostic trail.
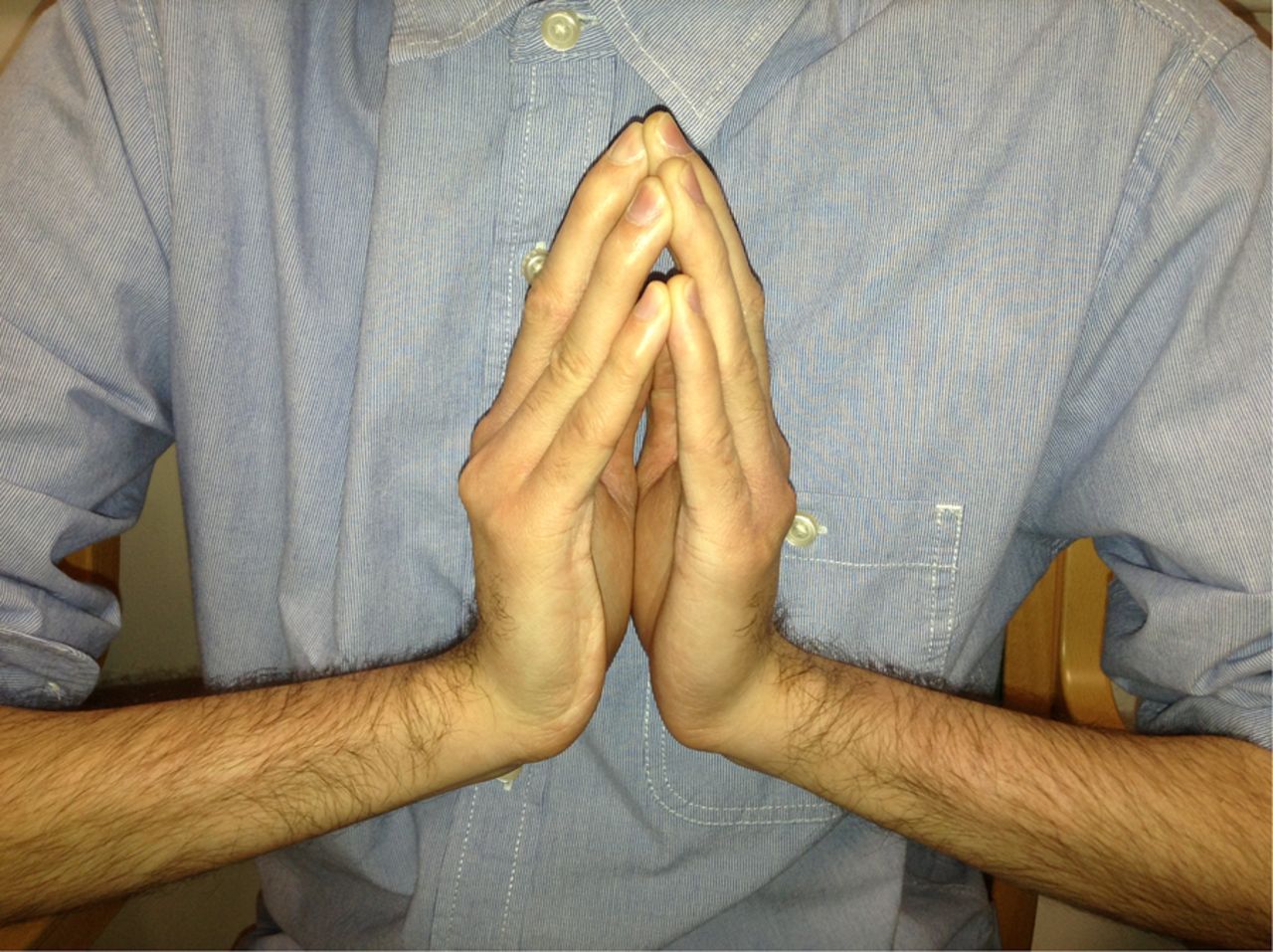
Prayer sign.

Fingers able to extend with the wrist in a neutral position.

Fingers cannot extend when the wrist is extended.

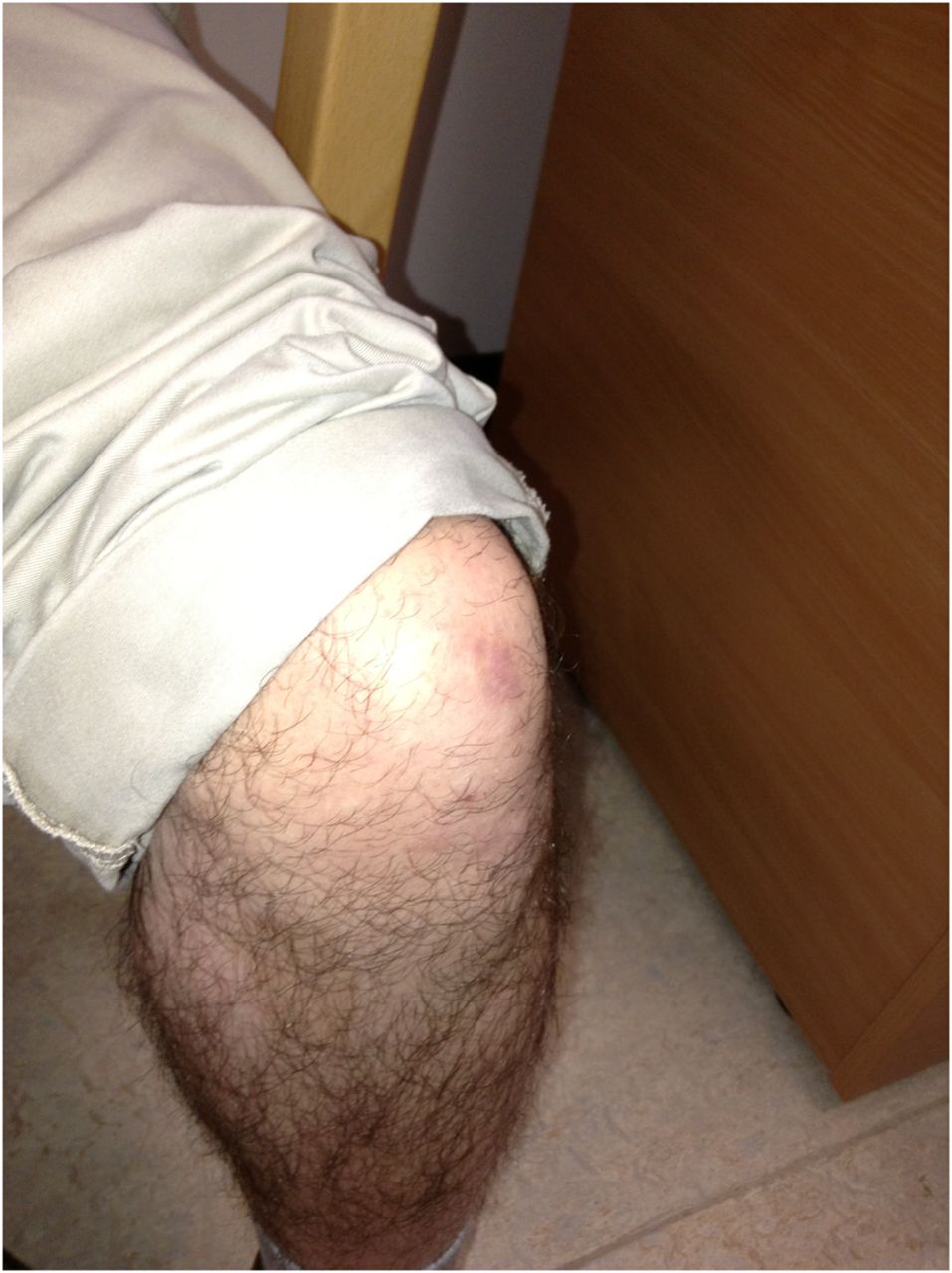
Ankle contractures.
The term ‘contracture’ has two meanings in muscle disease. It can refer, as in this instance, to an inability to stretch a muscle passively to its proper length because of fibrosis. Such contractures tend to develop late in the natural history of many myopathies, coinciding with prolonged static positioning and increasing reliance on wheelchairs. Early and prominent contractures that develop when muscles are still relatively strong, on the other hand, occur only in a few myopathies and therefore the differential diagnosis narrows considerably, see boxes 1 and 2.
Collagen VI disorders and Emery–Dreifuss muscular dystrophy
Collagen VI-related disorders—(see patient 1 vignette):
Bethlem myopathy and Ullrich congenital muscular dystrophy caused by mutations in one of three genes encoding collagen VI (COL6A1, COL6A2 on chromosome 21q22.3 and COL6A3 on chromosome 2q37).
Bethlem myopathy (autosomal dominant) and Ullrich congenital muscular dystrophy are at opposite points on the collagen VI disorder spectrum, sometimes with considerable overlap.
Bethlem myopathy—see text—patient 1.
Ullrich congenital muscular dystrophy is usually recessively inherited and causes severe weakness, proximal joint contractures, hyperlaxity of distal small joints and often hip dislocation at birth. Children rarely walk independently for any significant period. The calcanei protrude posteriorly in the feet. Respiratory involvement is common with nocturnal respiratory support in the first or second decade.
Emery–Dreifuss muscular dystrophy (see patient 2 vignette):
X-linked recessive—mutations in STA gene (protein emerin)
Autosomal dominant—mutations in LMNA on chromosome 1; LMNA gene encodes both lamins A and C—by alternative splicing.
All these are nuclear membrane proteins, helping to maintain the structural integrity of the nucleus and protecting it from mechanical stress.
Joint contractures are often in the elbows, posterior cervical muscles and ankles. In X-linked Emery–Dreifuss muscular dystrophy (XL-EDMD) contractures are the first sign whereas in AD-linked EDMD (AD-EDMD) contractures may appear after the onset of weakness. Loss of ambulation can occur in AD-EDMD but is rare in XL-EDMD. Supraventricular and ventricular arrhythmias, disorders of atrioventricular conduction and dilated cardiomyopathy characterise the cardiac manifestations; there is a high risk of cerebral emboli as well as sudden cardiac death, particularly in AD-EDMD.1
Emery–Dreifuss phenotype can also be caused by:
FHL1 gene (four-and-a-half-LIM)
SYNE-1 (synaptic nuclear envelope protein 1 or nesprin)
SYNE-2, TMEM43 (transmembrane protein 43, encodes LUMA)
Other neuromuscular conditions associated with rigid spine or other contractures (spinal rigidity characterised by limitation in flexibility of cervical, thoracic or lumbosacral spine)
FHL1-related genetic conditions and other myofibrillar myopathies:
Myofibrillar myopathies are genetically and clinically heterogeneous but all have characteristic histological features, including disintegration of myofibrils that begins at the Z-disc. They present, most often, between 25 and 45 years old with a proximal, distal or scapuloperoneal distribution of weakness and extraskeletal manifestations that include cardiac arrhythmias, cardiomyopathy, smooth muscle problems such as gastrointestinal pseudo-obstruction, respiratory muscle weakness and contractures. Contractures notably occur in FHL1, BAG3 and myotilin-related myofibrillar myopathies. FHL1 causes several different syndromes, but spinal rigidity, scapular winging, contractures, respiratory muscle weakness and cardiomyopathy are common features.
Some types of congenital myopathies:
These tend to present at birth or early infancy and are characterised by muscle weakness and specific structural abnormalities on muscle biopsy, can occasionally be associated with spinal rigidity:
Selenoprotein-related myopathy (SEPN1). SEPN1 causes a range of conditions, often there are multi-minicores on muscle biopsy; it also causes a congenital muscular dystrophy, rigid spine muscular dystrophy. People with SEPN1-related myopathy often remain ambulant well into adult life but have significant respiratory weakness.
Patients with centronuclear and nemaline myopathies also occasionally develop spinal rigidity.2
Rigid spine occasionally occurs in acid maltase deficiency.3
Limb girdle muscular dystrophy 2A:
Can have prominent contractures—including ankles and elbows. There is generally sparing of respiratory muscles, symmetrical scapular winging, and weakness particularly in hip adductors, elbow flexors, glutaeus maximus and abdominal weakness.
Other causes that present in early life:
Congenital muscular dystrophies—heterogeneous, mainly autosomal-recessive group of disorders, with dystrophic changes on muscle biopsy presenting at birth or within first few months of life. The weakness is often static but as contractures and scoliosis worsen so too does disability. Some affected people have intellectual problems with structural brain abnormalities. The collagen VI disorder, Ullrich congenital muscular dystrophy, is a type of congenital muscular dystrophy, as is rigid spine muscular dystrophy.
Congenital autosomal-recessive spinal muscular atrophy
Schwartz–Jampel syndrome (chondrodystrophic myotonia)
Congenital myasthenia gravis
The term ‘contracture’ in muscle disease can also refer to the sustained painful muscle contraction, precipitated by exercise, lasting up to hours, that characterises glycolytic metabolic myopathies, such as McArdle's disease. These are electrophysiologically silent, unlike the banal muscle cramps we all experience, and their presence is one feature that helps to distinguish glycolytic disorders from lipid metabolism disorders, such as carnitine palmitoyltransferase II deficiency, in which such contractures do not occur.4 ,5
The first patient described in the vignette has Bethlem myopathy with long finger flexor contractures, ‘the prayer sign’ and inability to extend fingers when the wrist is extended (figures 1⇑–3). In the right context this prayer sign strongly suggests Bethlem myopathy. There were contractures at the ankles (figure 4) and elbows too. He also had follicular hyperkeratosis (figure 5) and ‘cigarette paper’ scars (figure 6), both often occurring in Bethlem myopathy.

Follicular hyperkeratosis.
‘Cigarette paper’ scar.
Bethlem myopathy, first described in 1976 by Jaap Bethlem and George van Wijngaarden, is often referred to as ‘benign’ but approximately 10% of patients need nocturnal respiratory support and two-thirds require a walking aid after 50 years of age.4–6 Bethlem myopathy is a collagen VI-related disorder (see box) caused by mutations in one of three genes encoding collagen VI.
Cardiac involvement is uncommon. Muscle imaging can show several tell-tale features such as an abnormal signal in the centre of rectus femoris, a ‘central shadow’.4 ,5
Muscle histopathology in Bethlem myopathy ranges from a mild myopathy to more dystrophic changes and immunolabeling of collagen VI often shows surprisingly little abnormality in muscle samples. Collagen VI studies are more sensitive when applied to dermal fibroblast cultures from skin biopsy. Genetic testing remains the gold standard for diagnosing Bethlem myopathy, but it can be challenging to detect mutations because of the large size of the genes involved and therefore both imaging and skin biopsy remain potentially helpful diagnostic approaches before definitive genetic testing.4 ,5
In addition to Bethlem myopathy I would also like to highlight Emery–Dreifuss muscular dystrophy (EDMD) (box 1) as adult neurologists are likely to encounter these two conditions. There are two major forms of EDMD, an autosomal-dominant (AD-EDMD) and an X-linked (XL-EDMD) form.
Patient 2
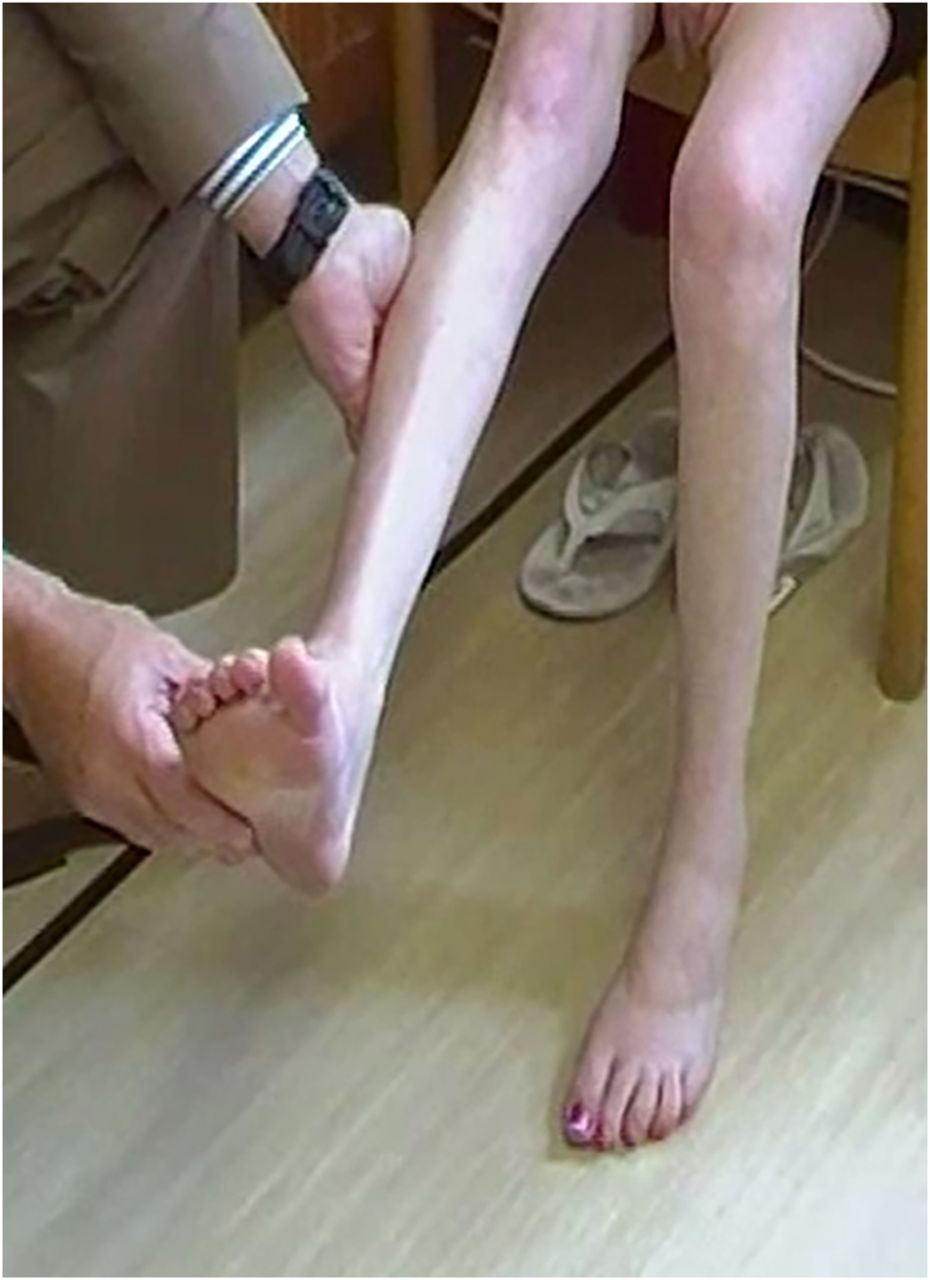
The woman in figures 7⇓⇓–10 has EDMD secondary to a mutation in LMNA gene (AD-EDMD). She walked on her toes as a young girl and was aware of weakness in her legs during her early teens. She had ankle contractures early in life and had several ankle operations and always found it very difficult to walk on bare feet. Although she loved playing netball it became increasingly difficult and from her mid-teens on, she was forced reluctantly to stop because she was simply too weak to compete. Despite this, she was a consummate expert at living the typical chaotic full-time life of a teenager.

Neck extensor contracture—unable to rest chin on chest.

Spinal contracture.

Humeral muscle atrophy with elbow contracture.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
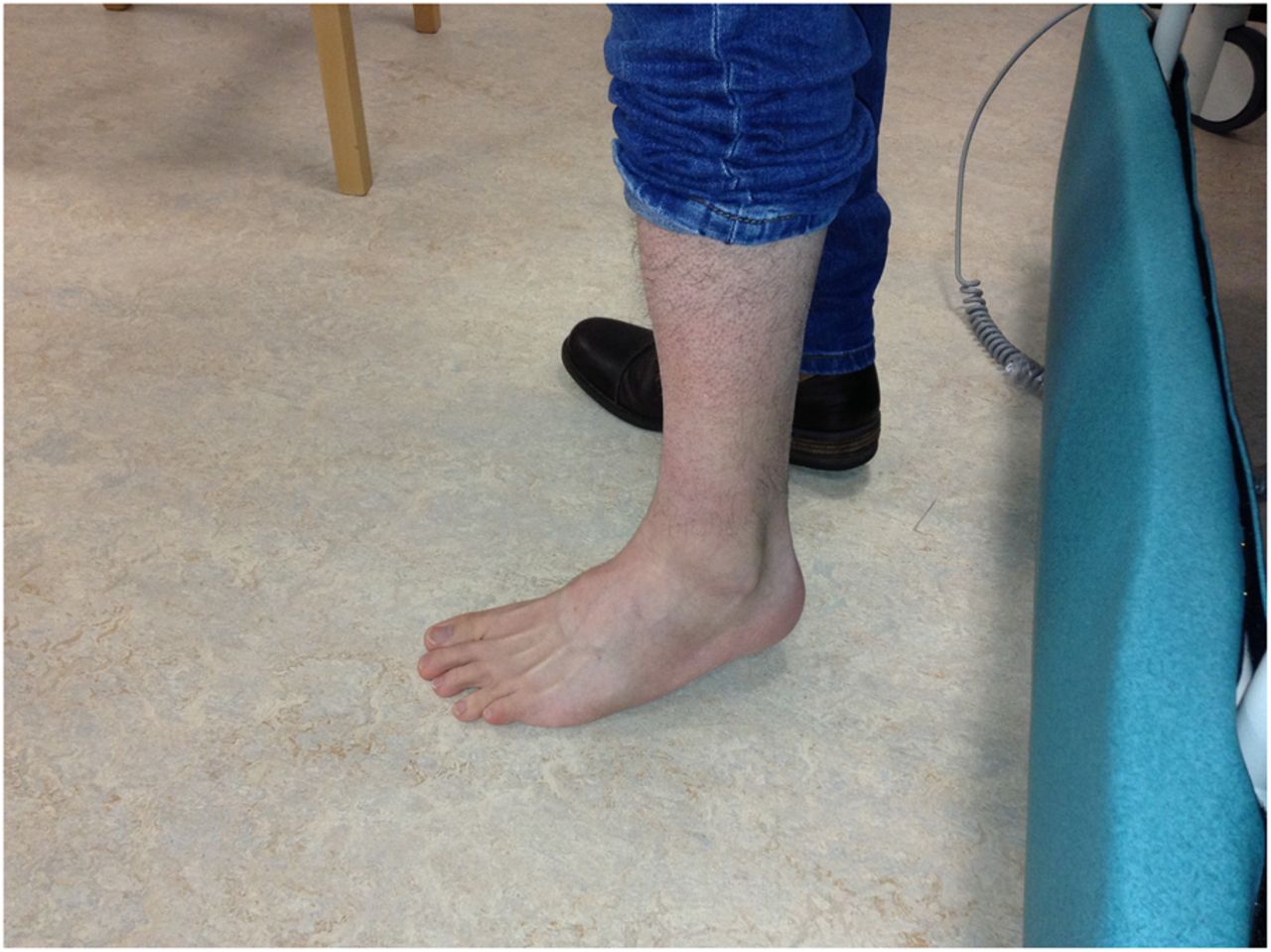
Wasting of the anterior tibial and peroneal muscles.
In her early 20s she fell and fractured her leg. She was given opiate pain relief in hospital while awaiting further orthopaedic assessment. That evening in hospital she had a respiratory arrest and was serendipitously found almost immediately and transferred to intensive care; perhaps the analgesia had precipitated a ‘house of cards’ moment.7 It soon became apparent that she was very weak, had contractures, was in atrial fibrillation and had significant respiratory muscle weakness. She is now on a nocturnal ventilator, has an implantable defibrillator and can stand but can take only a few steps.
Her neck extensor contractures mean she cannot rest her chin on her chest (figure 7). She also has spinal contractures (figure 8). She had contractures of the elbows and ankles but no long finger flexor contractures, in contrast to most people with Bethlem myopathy. She has atrophy and weakness of humeral muscles and peroneal muscles—a humeroperoneal distribution of weakness (figures 9 and 10). Although she has pes cavus with wasting of her tibialis anterior and peroneal muscles she also has hypertrophied extensor digitorum brevis muscles that help distinguish her distal muscle weakness from a longstanding neuropathy, such as Charcot–Marie–Tooth. People with AD-EDMD tend to have hypertrophy of extensor digitorum brevis, whereas in the XL-EDMD form extensor digitorum brevis hypertrophy is less prominent.
Her cardiac problems, humeroperoneal distribution of weakness and early contractures comprise the typical EDMD triad. As the natural history of EDMD unfolds, the pelvic girdle muscles also become weak. Patients may need a pacemaker and often a cardiac defibrillator to prevent sudden cardiac death.
There are several potential manifestations associated with mutations in the LMNA gene, collectively known as laminopathies.8 In addition to AD-EDMD the laminopathies can manifest as an autosomal-recessive axonal form of Charcot–Marie–Tooth disease, an autosomal-dominant proximal limb-girdle muscular dystrophy type 1B as well as non-neurological manifestations including partial lipodystrophy, a progeroid syndrome and isolated cardiomyopathy with conduction defects. There is sometimes overlap with, for example, an EDMD phenotype associated with fat accumulation in neck and face and loss of subcutaneous adipose tissue from the limbs and trunk as well as considerable intrafamilial and interfamilial variability for the same LMNA mutations. It is vital, therefore, to recognise the spectrum of laminopathies to enable prompt screening of at-risk family members. All first-degree relatives I think should be screened cardiologically, even if apparently asymptomatic.
A suspected clinical diagnosis of AD-EDMD can be confirmed by molecular genetic testing of LMNA; serum CK is normal to moderately elevated; EMG is myopathic and muscle biopsy shows non-specific myopathic or dystrophic changes. Patients with suspected XL-EDMD should have genetic testing of the STA gene (emerin gene); emerin is ubiquitous and therefore immunofluorescence or western blot can be undertaken in several tissues including skin.4 ,5
Box 2 summarises other neuromuscular conditions associated with contractures.
Summary
In summary, early contractures in patients can help guide you to a diagnosis of muscle disease; it is important to recognise EDMD in particular, as the potentially malevolent cardiac pathology can lead to sudden death, which is preventable with a pacemaker and often an implantable defibrillator.
Key points
Early prominent contractures are an important clinical clue in muscle disease as they dramatically refine the number of potential diagnostic possibilities.
Emery–Dreifuss muscular dystrophy comprises a triad that includes early muscle contractures—usually of the neck extensors, elbows and ankles—as well as early humeroperoneal weakness and potentially serious cardiac complications.
Cardiac complications of Emery–Dreifuss muscular dystrophy may include sudden cardiac death that is potentially preventable with an implantable cardiac defibrillator; placement of such devices needs consideration particularly in the autosomal-dominant form caused by LMNA mutation.
In collagen VI disorders (Bethlem myopathy) contractures develop particularly in the long finger flexors—an almost pathognomonic feature (prayer sign)—as well at the elbows and ankles; respiratory muscle weakness may develop but cardiac complications are rare.
Footnotes
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Commissioned; externally peer reviewed. This paper was reviewed by David Hilton-Jones, Oxford, UK.
Linked Articles
- Editors' commentary
Other content recommended for you
- Collagen VI related muscle disorders
- Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disorders
- Ullrich congenital muscular dystrophy: clinicopathological features, natural history and pathomechanism(s)
- Automated genomic sequence analysis of the three collagen VI genes: applications to Ullrich congenital muscular dystrophy and Bethlem myopathy
- Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes
- Targeted massively parallel sequencing and histological assessment of skeletal muscles for the molecular diagnosis of inherited muscle disorders
- Whole-genome sequencing and the clinician: a tale of two cities
- Rapidly progressive scoliosis and respiratory deterioration in Ullrich congenital muscular dystrophy
- Bethlem myopathy: long-term follow-up identifies COL6 mutations predicting severe clinical evolution
- Clinical spectrum and genetic variations of LMNA-related muscular dystrophies in a large cohort of Chinese patients