Article Text

Abstract
Skeletal muscle biopsy remains an important investigative tool in the diagnosis of a variety of muscle disorders. Traditionally, someone with a limb-girdle muscle weakness, myopathic changes on electrophysiology and raised serum creatine kinase (CK) would have a muscle biopsy. However, we are living through a genetics revolution, and so do all such patients still need a biopsy? When should we undertake a muscle biopsy in patients with a distal, scapuloperoneal or other patterns of muscle weakness? When should patients with myositis, rhabdomyolysis, myalgia, hyperCKaemia or a drug-related myopathy have a muscle biopsy? What does normal muscle histology look like and what changes occur in neurogenic and myopathic disorders? As with Kipling’s six honest serving men, we hope that by addressing these issues we can all become more confident about when to request a muscle biopsy and develop clearer insights into muscle pathology.
- MUSCLE DISEASE
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Before considering when you might request a muscle biopsy, it is worth considering how a biopsy is done and what can be learnt from it. Having an understanding of muscle pathology enables us to communicate with pathology colleagues and, when results are available, to translate muscle biopsy results for our patients’ benefit. We already recognise how such a similar investment helps our discussions with neuroradiology colleagues.
What is a muscle biopsy?
Muscle biopsy can be either open or closed (needle or punch) and both have their champions. A closed biopsy has the advantages of being more cosmetically appealing, minimally invasive and potentially yielding more than one specimen. The small sample size, however, creates challenges with handling and orientation; if pathology is patchy it may be missed.
An open biopsy is more invasive, but the operator can see the muscle, avoid tendinous insertions and can obtain an adequate specimen for biochemical studies, which is potentially important in mitochondrial disease, as we shall see. The person taking the biopsy needs adequate experience, skill and interest. In adult practice, open biopsy is generally favoured.
An open biopsy is undertaken in a sterile operating theatre. The skin and subcutaneous tissue are infiltrated with a local anaesthetic, avoiding the muscle. The incision is 4–6 cm along the long axis of the limb. The skin, subcutaneous fat and fascia overlying the muscle are dissected, and the biopsy is taken with muscle fibre fascicles running longitudinally. Complications are rare but include infection, delayed haematoma formation, muscle herniation through fascial defect and wound dehiscence.
The biopsy, usually about 15×10×5 mm in size, is laid on a non-absorbent card in a dry specimen container. The principle is to ‘snap freeze’ the muscle in isopentane cooled by liquid nitrogen (to approximately −160°C). This renders the sample in as life-like a state as possible without using a chemical fixative. ‘Snap freezing’ allows the tissue to be cut for diagnostic investigations and prevents the damage caused by formalin fixation or ice crystal formation (slow freezing) (contrast figures 1 with 2).

Normal adult skeletal muscle: subsarcolemmal located nuclei, average fibre size about 40–80 µm, surrounded by sparse endomysial connective tissue and visible perimysial septae (H&E stain, ×100).

Freezing artefacts: fine sarcoplasmic holes (caused by ice crystals) prevent examination of fibres (H&E, ×100).
Which muscles should be biopsied?
Select a muscle that is weak but can overcome gravity. Very weak muscles show marked loss of muscle fibres with fatty or connective tissue replacement and reveal no remnant of the underlying disease process: an ‘end-stage’ biopsy. Avoid muscles that have been injured, injected or sampled by neurophysiology within 6 months, as these situations can result in finding misleading inflammation.
A limited number of muscles are biopsied: in the upper limb, biceps brachii or deltoid; in the lower limb, quadriceps femoris (vastus lateralis or rectus femoris) or gastrocnemius. The pathologist needs to know what these muscles look like when healthy. Fibre-type composition and the normal range of fibre size will vary between muscles, depending upon their functional role. If contemplating using another muscle, consult with pathology colleagues.
Timely communication with the person undertaking the biopsy and pathologist is key, including clinical details and our diagnostic suspicions.
What does normal muscle look like?
A transverse section of normal muscle reveals fibres of roughly the same size, each comprising hundreds of myofibrils. The intermyofibrillar network sits between the myofibrils, composed of sarcoplasm, mitochondria and sarcoplasmic reticulum with the transverse tubular system. Nuclei occupy the periphery of the fibres (figure 1).
Each fibre is roughly polygonal transversely and packed snuggly alongside its neighbours, with the endomysium squeezed between adjacent fibres. Perimysium binds groups of fibres together, and the fascicles are collected together within the muscle belly, wrapped by the epimysium.
What changes do you see in denervated muscle?
Muscle fibres that have lost their nerve supply atrophy and become small angulated remnants, squeezed between their normal-sized, innervated neighbours. Denervated muscle fibres attract nerve twigs from healthy neighbours; the nerve twig sprouts and reinnervates the neurone-deprived myofibre. The reinnervated fibre assumes the same identity (type 1 or type 2 fibre) as its neighbour, since the reinnervating anterior horn cell exclusively innervates either type 1 or type 2 fibres. Reinnervated neighbouring muscle fibres, therefore, assume the same identity, and the normal mosaic distribution of fibre types becomes distorted into sheets of neighbouring fibres that are all the same type (fibre-type grouping).
What sort of changes do you see in myopathies?
In many myopathies, muscle fibre size is no longer uniform, and instead, there is considerable fibre size variation. Large fibres may split, particularly in muscle dystrophies. Nuclei, usually situated peripherally, start to migrate centrally.
Type 1 and 2 fibres may be selectively involved in some myopathies and in other health conditions. Type 2 fibre atrophy, for instance, typically occurs during immobility, corticosteroid use or general ill health. Type 1 atrophy occurs in some congenital myopathies; type 1 fibres tend to be more numerous than type 2 in congenital myopathies.
In many myopathies, the intermyofibrillar network changes appearance, becoming ‘moth-eaten’ and losing its characteristic regular cytoarchitecture.
The modified Gömöri-trichrome stain may show inclusions within muscle fibres, including the rod-like bodies in nemaline myopathy, or the darkly staining accumulation of myofibrillar material characteristic of myofibrillar myopathies (figures 3 and 4). In the core myopathies, oxidative stains such as nicotinamide adenine dinucleotide dehydrogenase (NADH)-TR may show well-demarcated areas of reduced or absent staining within the sarcoplasm (figure 5).

Nemaline myopathy: rod or granular red sarcoplasmic inclusions (Gömöri-trichrome stain ×400).
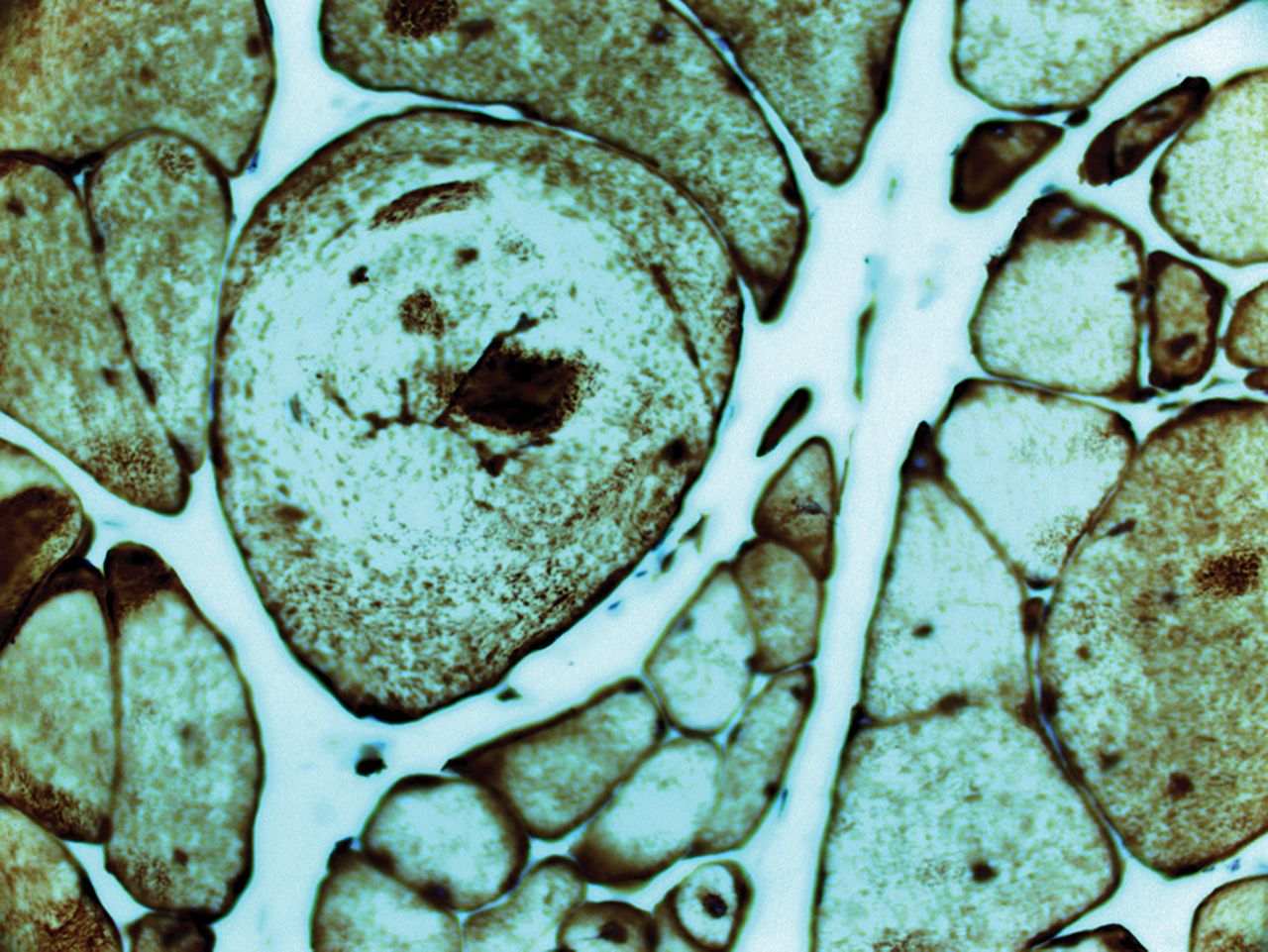
Myofibrillar myopathy showing marked fibre size variation, cytoplasmic inclusions and splitting fibres; desmin accumulation (desmin immunohistochemistry ×200).

Central cores: pale cores lacking in enzyme activity (nicotinamide adenine dinucleotide dehydrogenase stain).
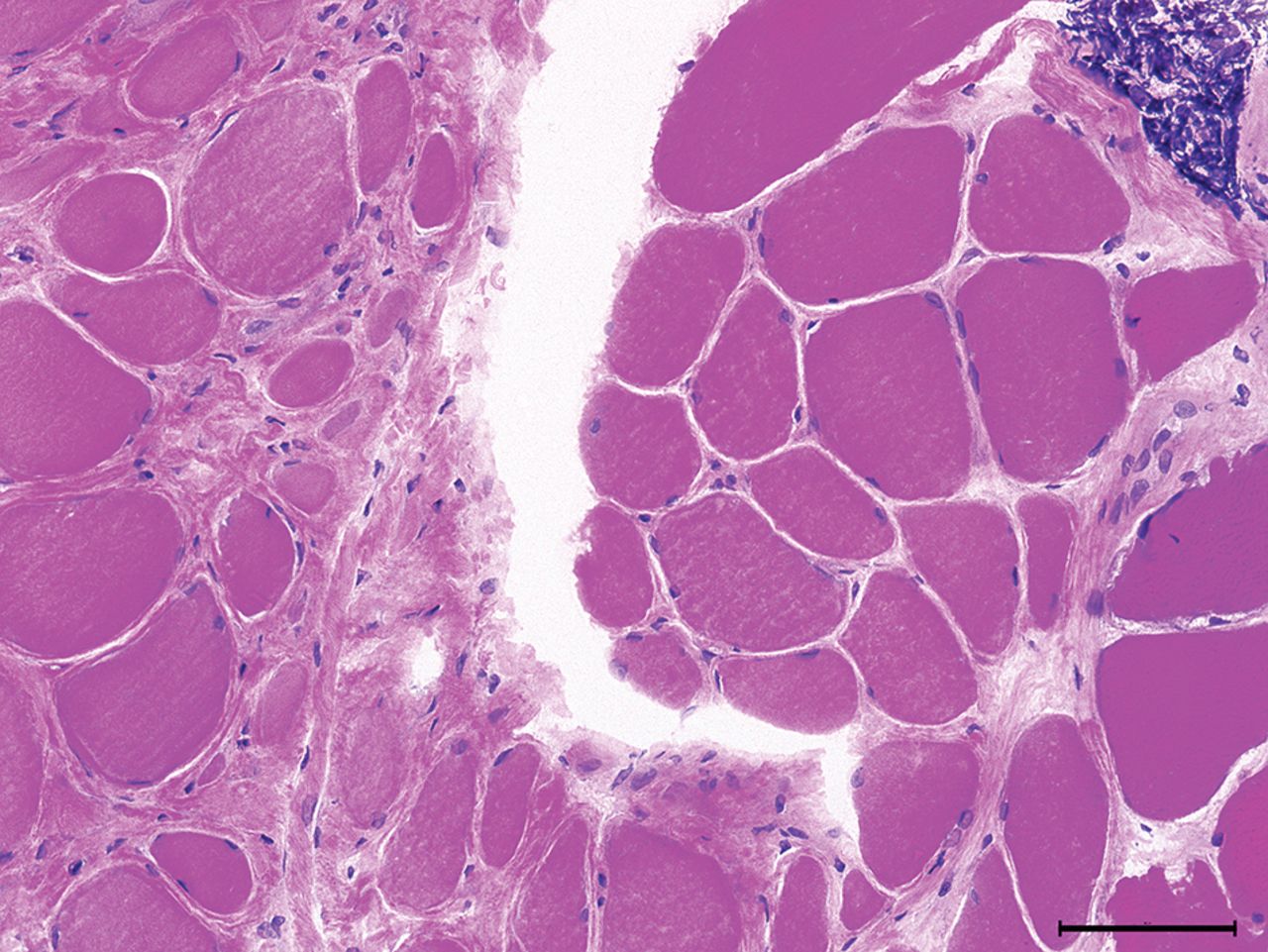
The endomysium becomes thickened and fibrotic in some muscle diseases and is particularly striking in muscle dystrophies (figure 6).

Limb-girdle muscular dystrophy: marked fibre atrophy, increased endomysial connective tissue suggesting dystrophy. A focus of lymphocytic infiltrate (top right corner) as sometimes seen in LGMD2B (H&E, ×200).
Inflammatory changes may develop in the endomysium of the inflammatory myopathies; immunohistochemistry can identify the types of inflammatory cell present (table 2). Immunohistochemistry can also identify the major histocompatibility antigen 1 (MHC1) on the sarcolemma in inflammatory myopathies or the membrane-attack complex deposit on the capillary endothelium in dermatomyositis. Inflammatory changes occasionally occur in genetic muscle diseases, including facioscapulohumeral dystrophy and dysferlinopathies (LGMD2B).
Western blot or immunoblot is a technique that uses gel electrophoresis to separate individual proteins from a mixture of proteins, according to their weight. Patients with Becker muscular dystrophy may have normal immunochemistry if the available antibodies happen to bind to the preserved parts of the dystrophin protein. Becker patients with smaller (80%) weight dystrophin on western blot have a genetic deletion, whereas those with a larger (5%) weight dystrophin have a duplication; the remainder have less of the normally sized dystrophin (15%).
No single pathological feature should be considered in isolation. Vacuoles in muscle fibres develop in some toxic myopathies, inclusion body myositis and some genetic myopathies. Focussing only on vacuoles might easily lead to mistaking a hydroxychloroquine-induced toxic myopathy for Pompe’s disease, since both have vacuoles.1 As the unassuming Miss Marple said, ‘One must provide an explanation for everything. Each thing has to be explained away satisfactorily. If you have a theory that fits every fact—well, then, it must be the right one’.
What stains are commonly used on muscle biopsy specimens and what do they tell us?
Tables 1 and 2 set out some of the stains commonly used and their contributions to understanding muscle physiology and pathology. In immunocytochemistry, the muscle biopsy is incubated with an antibody that is directed against a particular antigen.
Histology and histochemistry used on muscle biopsies
Examples of immunohistochemistry
In limb-girdle muscular dystrophies 2A and 2I, no immunocytochemical stain is invariably abnormal. Furthermore, since proteins interact with each other, there can be secondary immunocytochemical changes. For example, the transmembrane sarcoglycan complex links to the ‘C’ region of dystrophin; an abnormality in one sarcoglycan protein may disrupt its fellow sarcoglycans as well as dystrophin (all for one and one for all).2 3
Electron microscopy often provides further information, including identifying filamentous inclusions in inclusion body myositis or various abnormalities in congenital myopathies. Electron microscopy led to the discovery that mitochondria have their own DNA and that people with mitochondrial diseases have visible paracrystalline ‘parking lot’ inclusions within their mitochondria.4
When to request a muscle biopsy in a patient with suspected muscle weakness?
Investigating potentially myopathic weakness often requires consideration of a muscle biopsy. The practical neurologist aims to narrow the differential diagnosis clinically before embarking on investigations. There are many clues to help to distinguish between an acquired myopathy and genetic disorder, see further reading.35–7
Several myopathies may then become recognisable (or almost recognisable) at the bedside, and so the next diagnostic step might be a genetic or other blood test, rather than a muscle biopsy (table 3).
Some myopathies that may not require a biopsy for diagnosis
Gestalt impressions are vital in endocrine-related myopathies, confirmed with appropriate biochemical investigations (table 3). A muscle biopsy might still be necessary if there is no improvement despite restoring a normal hormonal milieu.
There are six patterns of weakness commonly referred to in muscle diseases; we look at each in turn and provide some guidance about the role of muscle biopsy in each. For the sake of this discussion, we have combined distal and scapuloperoneal forms.
Limb-girdle weakness
Generally, in a patient with a likely genetic cause for limb-girdle weakness, especially with pseudohypertrophy and raised serum creatine kinase, we request a dried blood spot test for α-glucosidase, and genetic testing for dystrophinopathy and LGMD2I, before a muscle biopsy.5 6 8 The clinical rules distinguishing genetic and acquired myopathies are robust but fallible, one reason to favour biopsy at this point.
However, practice varies, and some neurologists use next-generation sequencing before muscle biopsy. A muscle biopsy and next-generation sequencing are not mutually exclusive; patients often need both. Whole-exome sequencing, for instance, may identify large numbers of potential pathogenic variants. A muscle biopsy can help to identify an ‘in-depth phenotype’, steering us towards the culprit gene that is known to produce the particular muscle pathology.9 10
Distal or scapuloperoneal patterns of weakness
Muscle conditions with distal or scapuloperoneal patterns of weakness are also sometimes recognisable at the bedside (table 3). Myofibrillar or rimmed vacuoles are to be expected, but there may be other less common pathology; this favours an early biopsy rather than a narrower ‘phenotype-driven’ gene panel approach. For example, Cori–Forbes disease with its characteristic pools of subsarcolemmal glycogen can present with distal weakness.3 7 11
Distal arm and proximal leg weakness
This pattern raises suspicion of inclusion body myositis, although myotonic dystrophy type 2 (DM2) can have a similar distribution of weakness. We have a low threshold for testing for DM2, particularly if there are familial cataracts.
We recommend a biopsy to confirm inclusion body myositis and to exclude granulomatous myositis, which has a similar distribution of weakness but, unlike inclusion body myositis, may respond to immunosuppression.
Light microscopy in inclusion body myositis should show endomyseal inflammation, cytotoxic T cells partially invading non-necrotic muscle fibres and rimmed vacuoles. All three features are not always present; rimmed vacuoles may be a late feature. There are often mitochondrial abnormalities including COX-negative fibres or ragged-red fibres (see below). Electron microscopy shows 15 nm tubulofilamentous inclusions in the cytoplasm or nucleus, and immunohistochemistry may identify ‘Alzheimer-characteristic proteins’ such as hyperphosphorylated tau. Finding partial invasion and mitochondrial abnormalities is particularly supportive of a clinical diagnosis of inclusion body myositis.12
Progressive external ophthalmoplegia and/or ptosis
It is worth considering myasthenia, dysthyroid eye disease or oculopharyngeal muscular dystrophy, which do not require a muscle biopsy. Mitochondrial disorders then form the majority of the remaining patients.
We look for two common genetic causes of mitochondrial disease, the A3243G mutation in the mitochondrial genome (mtDNA) in a urine sample and POLG, a nuclear gene encoding the catalytic subunit of DNA polymerase gamma, detectable in leucocyte DNA. Polymerase gamma is one of several nuclear genes required to maintain healthy mtDNA; POLG mutations cause secondary mtDNA mutations that accumulate with age.7
If these are negative, we undertake a muscle biopsy from quadriceps or deltoid; levator palpebrae or orbicularis oculi biopsies are potentially rich in pathology, but limb muscles seem to be adequate.
Cytochrome c oxidase (COX, complex IV) histochemistry is centre stage when assessing adults with suspected mitochondrial disease. It reflects complex IV activity in the respiratory chain, and three of its subunits are mitochondrial DNA encoded, see figure 7.

Mitochondria—both mtDNA and nuclear DNA contribute to the respiratory complexes. Nuclear encoded proteins are red and mtDNA-encoded proteins are green. Note complex II is made exclusively from nuclear encoded proteins.

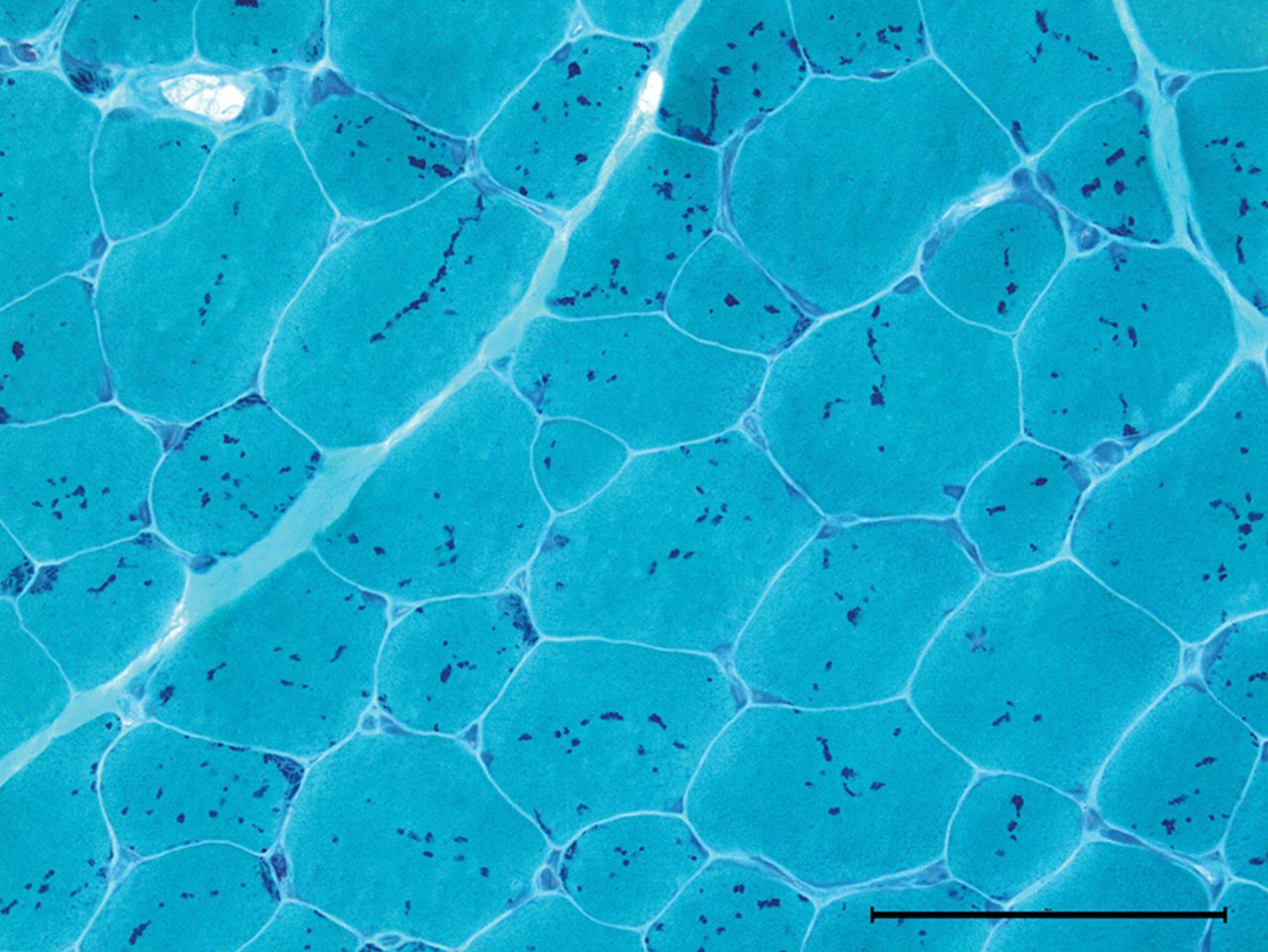
(A) COX-negative fibres take up the blue SDH stain but not the brown COX stain. The fibres should all stain brown, but fibres with higher levels of mitochondrial heteroplasmy appear blue among a sea of more normal brown fibres (COX-SDH stain). (B) Many COX-negative fibres and a ragged-blue fibre (left upper margin), evidence of a compensatory proliferation of mitochondria within a struggling myofibre. COX, cytochrome oxidase; SDH, succinate dehydrogenase.
There are multiple copies of mtDNA within each cell—the myofibre in skeletal muscle. Most frequently, mtDNA mutations affect only a proportion of mitochondria within each myofibre, termed heteroplasmy, and the fraction varies widely between myofibres.
Patients with mtDNA mutations, who account for approximately two-thirds of adult mitochondrial cases, therefore have a mosaic of ‘COX-negative’ fibres reflecting differences in heteroplasmy between myofibres. Myofibres with higher heteroplasmy tend to be COX negative. The remaining third of adult mitochondrial patients have nuclear DNA abnormities and often a more uniform reduction in COX staining, with secondary mtDNA abnormalities globally affecting all myofibres.7 13
COX-negative fibres are best demonstrated by serially staining for COX, followed by complex II (succinate dehydrogenase). Succinate dehydrogenase is entirely encoded by nuclear genes and therefore maintains normal activity irrespective of heteroplasmy levels, see figures 7 and 8.
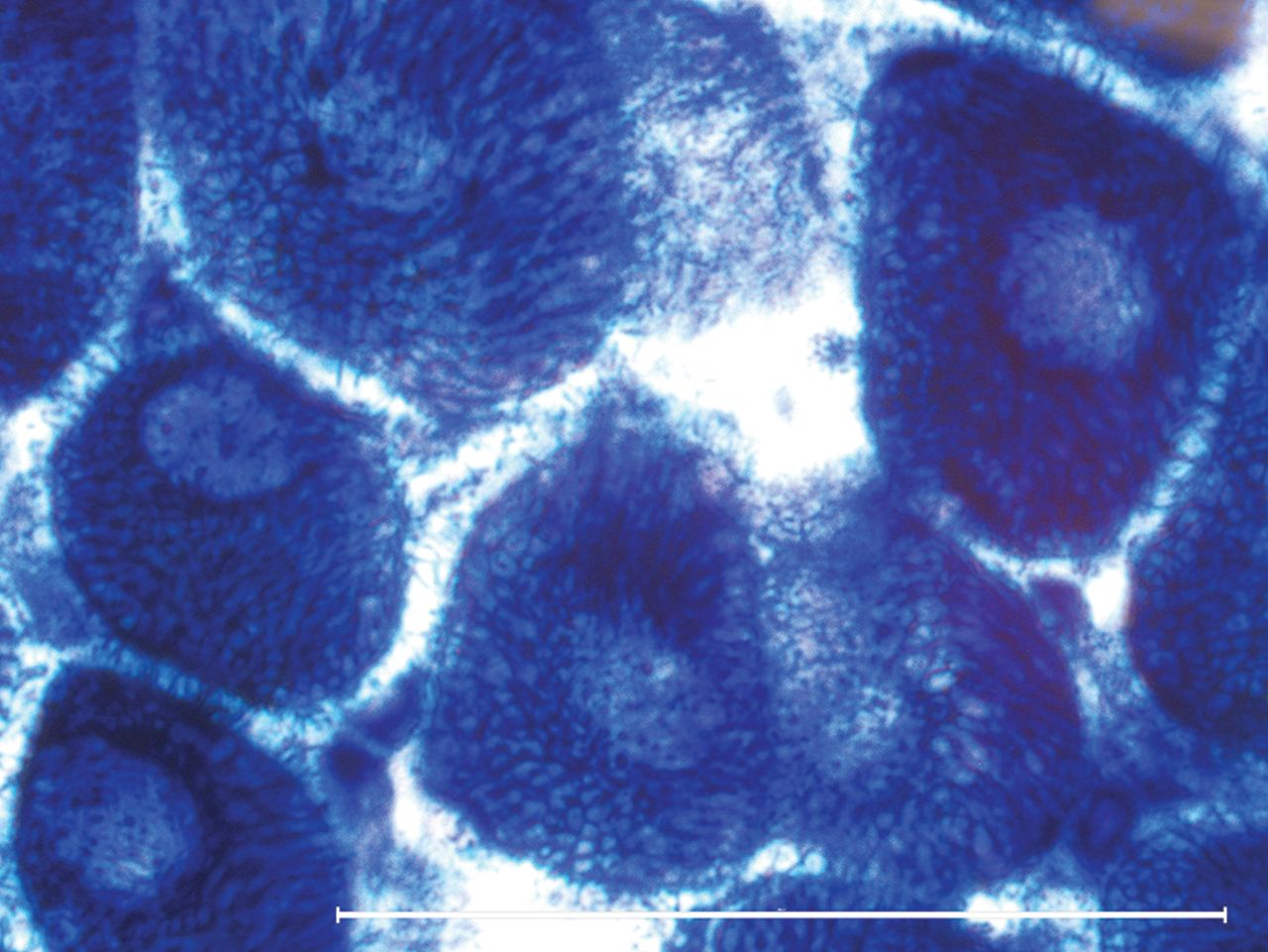
Myofibres with higher heteroplasmy may also show compensatory mitochondrial proliferation—the ‘ragged’ part in ragged-red or ragged-blue fibres (figures 8 and 9).

Ragged-red fibres, like ragged-blue succinate dehydrogenase stain, reveal mitochondrial proliferation in struggling myofibres (Gömöri-trichrome staining).
It is worth knowing that the A3243G mutation, the most common cause of mitochondrial disease, can have surprisingly well-preserved COX staining. A useful clue in this circumstance is the strong succinate dehydrogenase staining visible in the blood vessel walls of muscle.7 14 15
When histochemical clues are not forthcoming but suspicion remains, then respiratory chain enzyme analysis and/or quadruple immunofluorescence that quantifies complex I and IV in the muscle biopsy can help identify abnormalities.15–17
Muscle is also the gold standard tissue for examining mtDNA.18 In tissues with high replication rates, such as blood, heteroplasmy tends to decrease with age. Dividing cells with higher heteroplasmy may be ‘weeded out’ as they are at an ‘evolutionary’ disadvantage. Detecting the A3243G in a blood sample, therefore, declines with age, although it remains detectable in urinary epithelial cells.19 In postmitotic muscle, heteroplasmy remains stable or increases; for instance, mtDNA with a deletion might replicate faster than wild-type mtDNA. Our postmitotic skeletal muscle is therefore ideal for mtDNA genetic analysis.
It is a privilege to grow old with our mitochondria, but as we do so it is worth remembering that the proportion of COX-negative myofibres increases, to up to 5% over 50.20
Neck extensor weakness
We avoid biopsy of neck extensor muscles, as their normal histology is not well characterised. If weakness remains isolated to neck extensors, then we consider a period of 3–6 months of immunosuppression as a subset of patients with isolated neck extensor myopathy can respond.
Isolated neck extensor myopathy, myasthenia gravis (including antimuscle-specific kinase, MuSK antibodies) and motor neurone disease are the three most common disorders that may present with isolated or predominant neck extension weakness. Hyperthyroid and hyperparathyroid myopathies may also present like this.
Myotonic dystrophy, Pompe’s disease and facioscapulohumeral dystrophy may also develop neck extension weakness but frequently in advanced stages with weakness elsewhere including concurrent neck flexion weakness. These disorders require electrophysiology, serology and other blood tests but none generally requires a muscle biopsy.21
What if we suspect myositis?
We biopsy a weak muscle that can overcome gravity when we suspect an idiopathic inflammatory myopathy in the vast majority of cases. We simultaneously look for myositis-specific and myositis-associated antibodies. Myositis-specific antibody is not invariably present (about 60% of dermatomyositis cases) but provide additional insights into the likelihood of comorbidities, such as malignancy or interstitial lung disease, and likely response to treatment.22
Very occasionally false-negative biopsies do occur, since pathology can be patchy. In this situation, we organise MRI to highlight muscle oedema and pathology, revealed as high-signal intensity on short tau inversion recovery sequences to guide us to a biopsy site.23
Occasionally, if there are clear clinical clues, such as a typical dermatomyositis rash and only mild weakness, we rely solely on antibody testing initially without a biopsy, see case 1.
What is the role of biopsy in patients who have had rhabdomyolysis?
The acronym ‘RHABDO’ is a useful prompt to help identify those patients who may require further investigation, possibly including a biopsy (box 1).24 25 Delay the muscle biopsy for about 8 weeks following rhabdomyolysis, otherwise the overwhelming destruction will dwarf and obscure other diagnostic features.
RHABDO stands for
R—Recurrent episodes of exertional rhabdomyolysis
H—HyperCKaemia more than 8 weeks after event
A—Accustomed to exercise
B—Blood creatine kinase (CK) concentration above 50× upper limit of normal
D—Drug ingestion insufficient to explain exertional rhabdomyolysis
O—Other family members affected or other exertional symptoms
The causes of rhabdomyolysis are categorised as traumatic, non-traumatic exertional and non-traumatic non-exertional.
The non-traumatic exertional group includes the metabolic myopathies. Their workup often includes a gene panel for rhabdomyolysis and metabolic disorders—their clinical features are described elsewhere (see further reading).24 26 Some myopathies that cause non-traumatic exertional rhabdomyolysis are not included in such a panel, for example, muscle dystrophies (case 2), ryanodine receptor myopathies and mitochondrial disorders.27–29 Clinicians should seek relevant clinical clues when reviewing patients following their crisis. The indications for biopsy in these circumstances will then be similar to those when suspecting a dystrophy or a mitochondrial disorder (outlined above).
Rhabdomyolysis precipitated in someone taking statins when given medication that interferes with statin breakdown does not require a biopsy. However, a biopsy may be appropriate for someone taking statins who develops a suspected immune-mediated necrotising myopathy; such a complication can occur months to years after starting statins (see further reading).
What is the role of biopsy in patients with hyperCKaemia?
If a patient’s serum CK has been high, then repeat the test after refraining from exercise for a week. Interpretation requires flexibility; the heavyweight champion of the world will probably have a serum CK beyond the upper limit of normal.30
Drugs, endocrine, metabolic problems as well as macro CK can all raise the serum CK concentration. After considering these, many neurologists then proceed to electrophysiology and a possible biopsy. Next-generation sequencing can play a role, but once again, biopsy is often required for a ‘deep phenotype’ to help interpret the genetic tests.9
A diagnosis is more likely if electrophysiology shows myopathic changes, if the patient is young (<25 years), or if the serum CK is over three times the upper limit of normal. We tend to consider performing a muscle biopsy when a patient has one or more of these.31 We warn patients that the chances of a diagnosis are only about 1 in 4. These diagnoses may include metabolic myopathies, dystrophinopathies, limb-girdle muscular dystrophies and inflammatory myopathies.32 33
When should we consider a biopsy for a potential drug-related muscle complication?
Table 4 summarises some of the medications that have provoked muscle problems that we have encountered recently. We consider a muscle biopsy if there is no improvement 3–6 months after drug withdrawal, but sooner if the patient worsens.34–36 We might also consider a biopsy if there are several competing narratives. For example, consider a patient with a renal transplant and underlying connective tissue disorder who develops a gradually progressive proximal weakness. Is it the corticosteroid, the colchicine or the statin? Could it be a myositis? Muscle biopsy can help; vacuolar changes without inflammation would, for example, incriminate colchicine.
Iatrogenic myopathies witnessed in muscle clinic over the last few years
What is the role of biopsy in patients with myalgia?
Muscle pain is a near-universal experience. For instance, it accompanies infections ranging from chikungunya (meaning ‘to become contorted’ in Kimakonde) through to humdrum colds and ‘influenza.
Clinical criteria define conditions like fibromyalgia and chronic fatigue. They are ‘bread and butter’ for our primary care colleagues and expertly managed by them.
The following might prompt further investigations:
Weakness. Pain can create the appearance of weakness, often with a sudden reduction in effort. Watching a person walking on their toes or heels, hopping, or walking up-stairs is more revealing than just a couch-based assessment.37
Myopathic electrophysiology
HyperCKaemia (>2 to 3 times)
Exercise-induced pain (including a second wind)
Hypertrophy or muscle atrophy
Myotonic dystrophy type 2 may present almost exclusively with myalgia, and similarly, Parkinsonism is easily overlooked in this context.
We explain to patients that the chance of diagnosis is low, perhaps 1 in 20 biopsies, and that non-specific abnormalities are common.38–41 An excellent article on muscle biopsy remarked, ‘If the clinician cannot find something wrong it is highly unlikely that the tests will!’.42
A variety of pathologies may emerge, including metabolic myopathies, congenital myopathies, neurogenic disorders, mitochondrial disorders, myositis and muscular dystrophies.41 43
What is the role of biopsy in patients with exercise-induced muscle pain?
Common causes for exercise-induced myalgia include neurogenic or vascular claudication; amyloid myopathy can rarely cause claudication.44
The celebrated ‘lactate burn’ is well known, being familiar to genuine athletes and also a beloved exhortation roared out by commentators and armchair athletes as the home straight beckons. However, patients with McArdle’s disease suffer exercise-induced pain but produce no lactate; lactate probably does not fully deserve its epithet.
This group includes the metabolic myopathies: their clinical features and workup are described elsewhere.26 Remember that the metabolic gene panel is not exhaustive; if the panel is negative but clinical suspicion remains then a muscle biopsy may reveal, for example, a mutation in cytochrome b, the only mtDNA-encoded gene in complex III.45
Other exercise-induced muscle phenomena, such as myotonia, often require electrophysiology and genetic tests rather than biopsy. Muscle biopsy has no role in suspected skeletal muscle channelopathy, for instance, and the diagnosis is genetic.
Some other potential indications for a muscle biopsy
Patients with suspected peripheral nerve vasculitis should have combined nerve and muscle biopsy since the chances of positive histology increase from about 50% to 70% compared with a nerve biopsy alone.46 A generous biopsy from an asymptomatic muscle may reveal non-caseating granulomas, in a challenging case of possible neurosarcoidosis.47 Early skin or muscle biopsy may help in the frequently elusive diagnosis of intravascular lymphoma.48
Case 1
A 36-year-old woman said her hands felt stiff in the morning, waking her from sleep. I (RJW) considered carpal tunnel syndrome, but nerve conduction tests were normal. At review, I noticed roughened hyperkeratotic skin on the radial border of her thumbs and on the ulnar border of her index fingers (figures 10). She had a mild proximal weakness that I had overlooked initially. Her serum CK was 2700 IU/L (25–200). The stiffness in her hands reflected a non-erosive arthritis.

(A and B) Mechanic’s hands.
She had positive anti-Jo-1 antibodies, one of the myositis-specific anti-aminoacyl transfer RNA synthetase antibodies. She had an antisynthetase syndrome with mechanic’s hands, myositis, arthralgia, Raynaud’s phenomenon and evidence of pulmonary fibrosis on high-resolution CT scan of the chest.
I did not undertake a muscle biopsy, the diagnosis seemed clear and she improved on corticosteroid and mycophenolate mofetil.
Case 2
A 22-year-old man with mild learning difficulty had experienced several bouts of rhabdomyolysis and myoglobinuria after exercise. For example, his forearms had become swollen and painful the day after activities such as go-kart racing and paint balling.
In his early teens, he had developed a malignant hyperthermia-type reaction after a general anaesthetic for wisdom tooth removal. A muscle biopsy after this had shown a positive in vitro contraction test. He was diagnosed with malignant hyperthermia. The biopsy was not analysed further and he had no genetic tests.
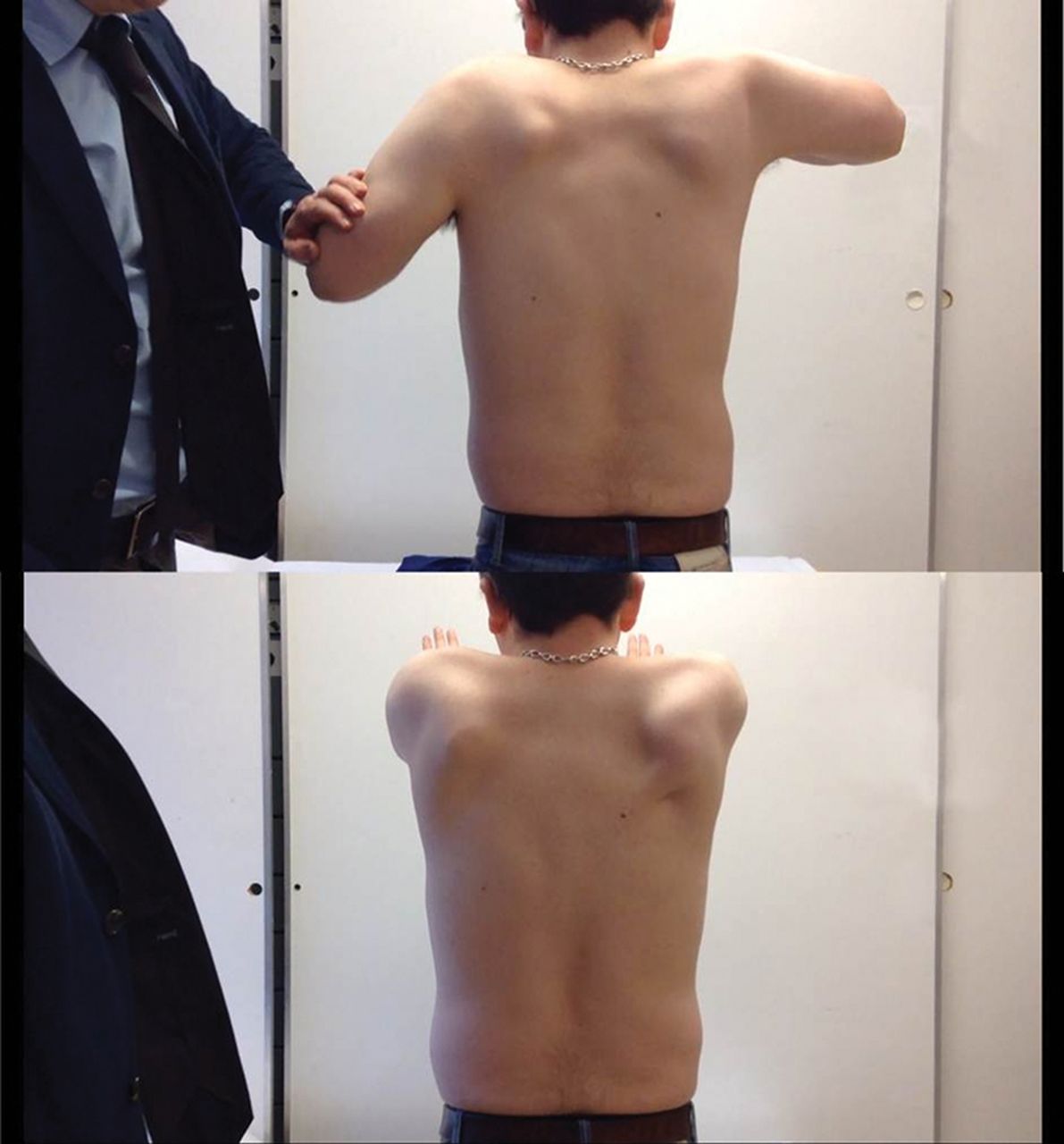
We considered a ryanodine mutation, given his malignant hyperthermia and exercise-induced rhabdomyolysis, but he had large calves, winged scapulae and mild limb-girdle pattern weakness (figure 11). His serum CK was about 1500 IU/L (25–200). Further immunohistochemical staining led to the diagnosis of Becker muscular dystrophy.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A and B) Person with Becker muscular dystrophy who has proximal shoulder weakness and scapular winging.
The practical message is that all muscle biopsies should be analysed methodically; clinicians and pathologists need to discuss each case. The diagnosis may have important implications for family members.
Key points
Muscle biopsy remains important in investigating potentially myopathic weakness, although its role differs with the pattern of weakness.
For myopathies recognisable at the bedside, a genetic or metabolic test may be the next step.
Muscle biopsy may help in myositis, rhabdomyolysis, hyperCKaemia, myalgia and drug-related myopathies, but only in a proportion of cases and after other investigations.
Clinicians should communicate early with those undertaking the biopsy and the pathologist, upon whose expertise we rely for interpretation.
Having some insights into muscle pathology enriches clinical life and helps us to decipher results for our patients.
Acknowledgments
We would like to thank Miss Georgia Robinson and Steve Atherton, Medical Illustration Department, Swansea Bay UHB for their considerable help and expertise.
Further reading
REFERENCES
Footnotes
Correction notice This article has been corrected since it was published Online First. An author name has been amended.
Contributors JW and AB contributed to the design, writing and review of this manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Consent obtained directly from patient(s).
Provenance and peer review Commissioned. Externally peer reviewed by Michael Rose, London, UK, and James Holt, Liverpool, UK.
Other content recommended for you
- Muscle disease
- Autoantibody testing in idiopathic inflammatory myopathies
- Inclusion body myositis: old and new concepts
- Muscle hypertrophy and pseudohypertrophy
- Muscle diseases: mimics and chameleons
- PYROXD1-associated myopathy
- Myopathy with antibodies to the signal recognition particle: clinical and pathological features
- DIAGNOSIS AND TREATMENT OF INFLAMMATORY MUSCLE DISEASES
- Clinical assessment determines the diagnosis of inclusion body myositis independently of pathological features
- Efficacy of immunosuppressive treatment in a systemic lupus erythematosus patient presenting with inclusion body myositis