Article Text

Statistics from Altmetric.com
A man in his mid-70s presented to an emergency department in Zambia with 2 months of progressive cognitive decline. His family reported that his problems began with an acute episode of non-sensical speech that had resolved within a few hours but with increased lethargy the next day. Within a month, he could no longer hold a coherent conversation about even basic events but remained independent in activities of daily living. His symptoms deteriorated over the next few weeks so that he required assistance with mobility, became increasingly uncooperative and disorientated, could not write his own name, was incontinent, resisted care, would get lost in his house and was no longer able to recognise his family. He had no delusions, hallucinations or headache. His medical history was notable for benign prostatic hyperplasia, type 2 diabetes mellitus and hypertension, which were all well controlled on medication.
At presentation, he was awake and alert, orientated to person only, confabulating and unable to identify family members. His speech was clear but tangential with reduced content. He was fluent with intact repetition. Naming was intact to high-frequency but not low-frequency words. He had ideomotor apraxia and a right homonymous hemianopia. The remaining cranial nerve examination was unremarkable, and motor examination was notable only for paratonia in all limbs. Tendon reflexes were normal, and plantar reflexes were flexor bilaterally. There were frontal release signs, including jaw jerk, palmomental, grasp and glabellar tap, but no startle reflex or myoclonus. Sensory, coordination and gait examinations were normal. He had no meningeal signs.
Questions for consideration
What is the likely localisation of his presentation?
What is the differential diagnosis?
His marked cognitive decline suggested diffuse cortical dysfunction. Frontal release signs suggested frontal lobe involvement while the episode of incoherent speech might suggest a focal seizure localising to Wernicke’s area on the left. Ideomotor apraxia localises to the premotor cortex in the dominant hemisphere, and the right homonymous hemianopia to the left hemisphere, either along the optic tract, at the lateral geniculate body, or in the calcarine cortex.
The rapid decline suggested a rapidly progressive dementia, with a wide differential diagnosis, including vascular (eg, multiple infarcts, thalamic or callosum infarcts, cerebral amyloid angiopathy), infective (eg, viral encephalitis, HIV dementia, urinary tract infections in an elderly person with mild dementia), toxic-metabolic (eg, vitamin B12 and B1 deficiencies, endocrine abnormalities, hyperglycaemia, metal toxicity), autoimmune (eg, central nervous system (CNS) lupus), malignancy/metastasis related (eg, carcinomatous meningitis, paraneoplastic encephalitis), iatrogenic (eg, illicit drug use, chemotherapy), neurodegenerative (eg, Alzheimer’s disease, prion disease, dementia with Lewy bodies) and systemic disease (eg, epilepsy).1 In case series from rapidly progressive dementia referral centres, common causes of rapidly progressive dementia include immune-mediated encephalopathies, prion diseases and neurodegenerative diseases such as Alzheimer’s disease and frontotemporal dementia.1 In other series, secondary reversible causes including infections were most common.2 Though the epidemiology in low-income settings is largely unknown, these settings might be expected to have higher rates of infective causes.
Creutzfeldt-Jakob disease (CJD) is the prototype of rapidly progressive dementia. Sporadic CJD, the most common form, tends to have early neurological signs. This patient’s visual field deficit did raise suspicion for CJD, although there was no startle response or myoclonus (common findings in CJD). We also considered infective causes of rapidly progressive dementia but these are often accompanied with systemic symptoms such as fever and meningism. Implicated microo-rganisms include viruses (eg, HIV, herpes simplex), fungal (eg, CNS aspergillosis), spirochaetes (eg, neurosyphilis) and bacteria (eg, CNS tuberculosis). This patient had no systemic symptoms to suggest an infective cause nor was he immunocompromised. Toxic-metabolic causes such as vitamin B12 deficiency should be considered, as should a personal history of exposure to toxins such as organic lead and mercury. However, there was no obvious history to suggest these possible causes.
Questions for consideration
What investigations might further evaluate this patient’s presentation?
Investigations for rapidly progressive dementia includes full blood count with differential; basic metabolic panel including calcium, magnesium and phosphate; HIV test; rapid plasma regain test; liver function and thyroid function tests; rheumatological screen including erythrocyte sedimentation rate, antinuclear antibodies and C reactive protein; urine for urinalysis, toxicology and culture; and cerebrospinal fluids for cell count with differential, protein, glucose, IgG index, oligoclonal bands, venereal disease research laboratory test, 14-3-3 protein, tau, cryptococcal antigen, viral PCRs, Gram stain and bacterial culture, fungal culture, acid-fast bacilli stains and cultures, cytology and flow cytometry. Imaging including MR scan of the brain with and without contrast and chest radiograph should also be obtained.1 Additional investigations are recommended in some clinical scenarios, including cancer screening, blood smear, homocysteine and additional rheumatologic tests such as anticytoplasmic antineutrophil cytoplasmic antibodies.
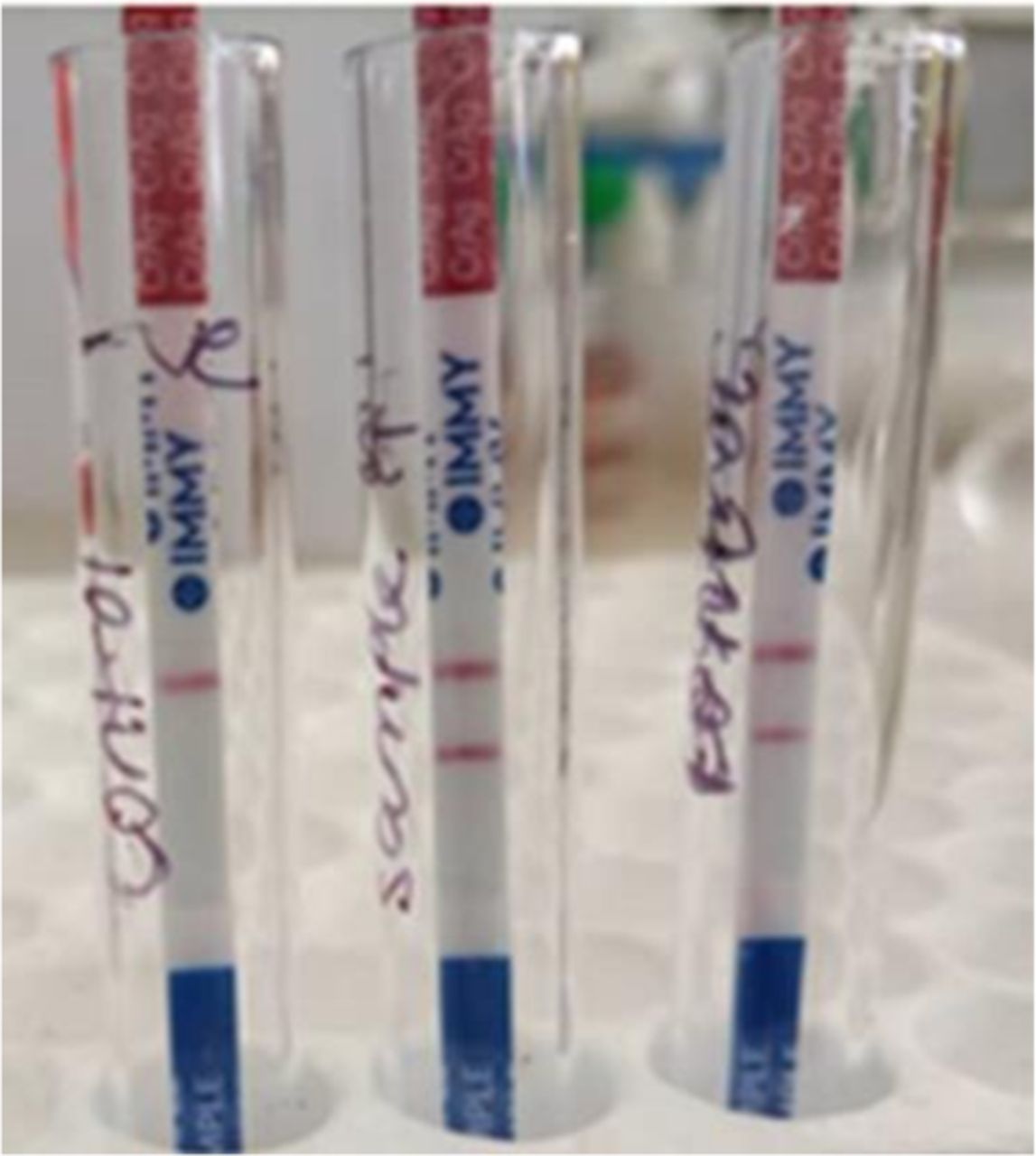
Given the resource limitations in our setting, the patient underwent a targeted and stepwise workup that included full blood count, metabolic panel and urinalysis, which were unremarkable. HIV ELISA and PCR were non-reactive. Chest radiograph showed bilateral nodular infiltrates with a small effusion in the left lower lung. MR scan of the brain showed an area of encephalomalacia in the left occipital lobe and generalised brain atrophy. Electroencephalogram (EEG) showed a slowed background rhythm without epileptiform discharges, consistent with moderate encephalopathy. Table 1 details his CSF studies and showed Cryptococcus spp (figure 1). The CSF opening pressure was not measured as there were no manometers available.
CSF findings
{kind=link}
This patient’s CSF cryptococcal antigen test result. The test was repeated twice to ensure the first unexpected result was not a false positive. Courtesy of University Teaching Hospital Microbiology Laboratory. CSF, cerebrospinal fluid.
We diagnosed cryptococcal meningitis and gave amphotericin B with high-dose fluconazole, because flucytosine, the preferred drug for use in combination with amphotericin B, is currently unavailable in Zambia. He developed complications with acute kidney injury, electrolyte imbalance and congestive heart failure, but these resolved with treatment. He was discharged home on oral fluconazole. His neurological symptoms improved after 3 weeks of treatment to the point where, he could recognise his family members, was no longer confabulating and had no ideomotor apraxia.
Discussion
Cryptococcal meningitis arises from an infection caused by the yeast Cryptococcus. The most common pathogens are Cryptococcus neoformans and Cryptococcus gattii, both of which occur naturally in decaying, organic material and in different types of soil, although with varying geographical distributions. Cryptococcal infection enters the body through inhaling fungal spores in soil contaminated by avian (usually pigeon) droppings, resulting in pulmonary cryptococcosis characterised by pulmonary micronodules and pleural effusions.3 These are typically asymptomatic in a non-immunocompromised host, as in this patient. Cryptococcal meningitis usually presents with a subacute to chronic meningitis or meningoencephalitis and typically affects people who are immunocompromised, especially those living with HIV.4 Cryptococcal meningitis is the most common cause of meningitis and one of the leading causes of death in people living with HIV in sub-Saharan Africa.5 ,6 For reasons that are not clear, most cryptococcal meningitis in people living with HIV is caused by C. neoformans while C. gattii is rare in this group but more common in people who are not immunocompromised. The cryptococcal antigen in this patient was detected using the CrAg lateral flow assay (Immy, Norman, Oklahoma, USA), which cannot differentiate cryptococcal species. The fungus also grew on Sabouraud’s dextrose agar, but we could not complete further speciation due to unavailability of media to identify up to species level in our laboratory at the time.
Various studies have shown that the epidemiology, risk factors, presentations and outcomes of cryptococcal meningitis vary between people living with HIV and those without HIV infection. People without HIV infection comprise a heterogenous group that includes recipients of solid organ transplants, people taking immunosuppressive medications, those with malignancies and those with poorly controlled diabetes mellitus. In addition to these groups are healthy individuals with no obvious risk for cryptococcal meningitis who make up a widely variable proportion of patients with cryptococcal meningitis, between 8% and 81% of patients with cryptococcosis in various centres.7 ,8 This group probably includes individuals with undetected innate immune deficiencies or a congruence of innate and acquired immunodeficiencies. However, new risk factors have emerged including recreational intravenous drug use and prolonged influenza virus infection.9 Both C. neoformans and C. gattii may cause cryptococcal meningitis in immunocompetent hosts, but C. gattii characteristically affects this group, accounting for more than 40% of infections in one study.7
CNS cryptococcosis typically presents as a subacute meningoencephalitis in most of both people living with HIV and HIV-uninfected individuals with features including meningism, fever, headache, focal neurological deficits and vomiting. However, cryptococcal meningitis rarely presents as rapidly progressive dementia and mimicking traditional dementia syndromes such as Alzheimer’s disease and vascular dementia.10 ,11 As such, cryptococcal meningitis may be misdiagnosed as these non-infective syndromes.
Rapidly progressive dementia has many causes that are often reversible, and patients need extensive diagnostic testing to identify treatable causes. However, in resource-constrained settings with limited diagnostic capabilities, establishing a definitive diagnosis requires a prioritised stepwise diagnostic approach focused on identifying potentially reversible causes. It is also important in the local setting to have a comprehensive understanding of the most common causes of rapidly progressive dementia, as causes differ by region, with infections likely being more common in low-resource settings such as Zambia. Therefore, it is important to exclude infective processes early in the diagnostic workup.
Key points
Cryptococcal meningitis may present as rapidly progressive dementia in people without HIV.
Where resources are limited, workup for rapidly progressive dementia should proceed in a prioritised, stepwise fashion based on prevailing local epidemiology.
Identifying reversible causes of rapidly progressive dementia remains the priority, including in settings with resource limitations.
Infective causes of rapidly progressive dementia may be more common in regions where CNS infections are prevalent, even in people with normal immunity.
Ethics statements
Patient consent for publication
Footnotes
Twitter @Chishimb@chishimba22, @mashina_chomba
Contributors LCC wrote the first draft. Review and editing, from the first to the final draft, was done by LCC, DS, SF, MA, SZ, MC, MM, KY-Z and KS. Microbiology and visualisation were performed by RN and SF. Clinical Management of the patient, including case conference discussions for management of the patient, were performed by LCC, DS, KY-Z, MM, KS, SF, MC, SZ and MA.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed by William Howlett, Tanzania, Africa.
Linked Articles
Other content recommended for you
- Patients with Alzheimer's disease and dementia with Lewy bodies mistaken for Creutzfeldt-Jakob disease
- Presenile dementia syndromes: an update on taxonomy and diagnosis
- Neuropathological investigation of dementia: a guide for neurologists
- The clinical assessment of the patient with early dementia
- Dementia
- Molecular pathology of neurodegenerative diseases: principles and practice
- Pathology of neurodegenerative disease for the general neurologist
- Rare histotype of sporadic Creutzfeldt-Jakob disease, clinically suspected as corticobasal degeneration
- Impaired imitation of gestures in mild dementia: comparison of dementia with Lewy bodies, Alzheimer's disease and vascular dementia
- A comparative analysis of cognitive profiles and white-matter alterations using voxel-based diffusion tensor imaging between patients with Parkinson's disease dementia and dementia with Lewy bodies