Article Text

Statistics from Altmetric.com
Case presentation (Dr RM Ahmed)
History
A 76-year-old man, a university educated retired engineer, was referred with a 2-month history of deteriorating balance and memory. He took metformin for type 2 diabetes mellitus. He attended with his wife within 2 days of the referral and brought his recent MRI scans of the brain and abdomen. He was not concerned by any memory problems (his wife certainly was), but felt off-balance when walking. It became apparent that he had actually been losing balance and memory for about 12 months, but this had progressed more rapidly in the last 2 months. He had no headaches, sensory or urinary symptoms. He was sleeping and eating well and had not lost weight. He was an accomplished pianist and his musical abilities had not deteriorated. He had never smoked and drank little alcohol.
Examination
His wife said that he was his usual jocular self—possibly even fatuous. General examination, including heart, chest and abdomen, was normal. There was no cranial nerve abnormality, no papilloedema, no muscle wasting, normal muscle tone and strength throughout; reflexes were normal in the upper limbs, but knee jerks and ankle jerks were absent. His right plantar was extensor. Sensation was intact to pinprick; there was slight impairment of vibration and position sense at the toes. He walked with a wide-based unsteady gait and could not tandem walk; Romberg's test was positive. He scored 22/30 on the Mini-Mental State Examination, losing points for orientation, memory (recall) and repetition. On the Addenbrooke's Cognitive Examination he scored 62/100, losing points on memory (recall, anterograde and retrograde). Verbal fluency was decreased. Language, naming and comprehension were intact. His scores were: attention and orientation 14/18, memory 10/26, fluency 0/14, language 24/26, visuospatial 14/16.
The MRI scan of brain (figure 1) he brought with him had been reported as follows: ‘There is moderate cerebral atrophy with prominence of the cortical sulci and of the ventricular system with dilatation of the bodies and temporal horns of the lateral ventricles and of the third ventricle. The fourth ventricle is prominent. There is increased signal intensity in the peri-ventricular white matter suggesting periventricular cerebrospinal fluid (CSF) resorption. There is a CSF collection in the cisterna magna deforming the inferior aspects of the cerebellar hemispheres, suggesting an arachnoid cyst. The images show no small vessel ischaemia. The distal left vertebral artery is occluded. Comment: despite the cerebral atrophy, the appearances suggest normal pressure hydrocephalus.’

MRI scan of brain at presentation. Axial T2 (A, B) and coronal fluid attenuated inversion recovery sequences show cortical atrophy and ventricular enlargement with possible periventricular cerebrospinal fluid exudation.
The abdominal MRI report described ‘four cysts in the pancreas, without enhancement, mass effect, surrounding infiltration or duct dilatation’.
Discussion Part A (Dr GM Halmagyi)
This patient has three distinct clinical syndromes: dementia (impaired memory and fluency), ataxia (wide-based gait and positive Romberg's test) and sensory neuropathy (absent knee and ankle reflexes with distal sensory impairment). The ataxia might be from sensory impairment, cortical impairment or an unrelated cause such as cerebellar or vestibular impairment. The tempo of the dementia and of the ataxia is subacute; the tempo of the neuropathy is unknown. Box 1 details the possible diagnosis to consider. What single disease could cause all three of these syndromes? Possible conditions include vitamin B1 deficiency (Wernicke–Korsakoff), vitamin B12 deficiency (subacute combined degeneration of the cord) and paraneoplastic disease.
-
Vitamin B12 deficiency would explain all the neurological problems1 ,2 and a low serum B12 would clinch the diagnosis, even without haematologic changes. Neurological B12 deficiency can occur even with normal serum B12 levels,3 particularly in patients exposed to metformin4 or nitrous oxide (medical or recreational).5 In such cases, high serum levels of homocysteine or methylmalonic acid—B12 intermediary metabolites—indicate functional B12 deficiency.6 Brain MRI changes can occur in B12 deficiency3 although spinal MRI changes are more usual.
-
Wernicke–Korsakoff syndrome, in the context of chronic alcohol abuse (to explain the peripheral neuropathy),7 would also explain all the neurological abnormalities, even without ophthalmoplegia. However, the patient was well nourished and drank little alcohol, making this diagnosis unlikely. Wernicke's encephalopathy has characteristic MRI abnormalities in the brainstem and the basal ganglia, including T2 hyperintensity around the third ventricle, in the dorsomedial regions of the thalami, the periaqueductal areas of the midbrain, and the mamillary bodies.8
-
Paraneoplastic disorders. Paraneoplastic limbic encephalitis typically presents with cognitive changes, including short-term memory impairment, seizures, behavioural and personality changes;9 there may be antibodies to Hu, CV2, Ma2 or voltage-gated potassium channels (VGKC).9 Anti-Hu is also associated with paraneoplastic cerebellar degeneration and a sensory neuropathy,10 which could explain this patient's gait disturbance. On MRI, the mesial temporal structures may be swollen and hyperintense on T2-weighted sequences.11 This patient had no behavioural and personality changes, psychiatric symptoms or seizures, and the lack of typical MRI abnormalities makes limbic encephalitis less likely.12
To investigate the possibility of limbic encephalitis further, we would need CSF, looking for mild leukocytosis and elevated protein.9 EEG could show focal or generalised slowing or even epileptiform activity maximal in the temporal regions.12 We also need to test for antineuronal antibodies and VGKC antibodies. He then needs imaging looking for an underlying tumour including a chest x-ray, whole body CT/positron emission tomography (PET) scan and also serum tumour markers (carcinoembryonic antigen (CEA), Ca19.9, α-fetoprotein).
The differential diagnosis for this patient includes
-
Hydrocephalus: communicating (including normal pressure hydrocephalus, or as a result of infection, infiltration of meninges), non-communicating.
-
Neoplastic condition: intraparenchymal and/or meningitic.
-
Paraneoplastic condition: for instance, anti-Hu antibody-producing neoplasm causing cognitive deficit and ataxia, or voltage-gated potassium channels and anti-NMDA receptor encephalitis.
-
Infective encephalitis: for instance, viral encephalitis, infective meningitis (TB, cryptococcal), HIV encephalitis, Whipple's, syphilis.
-
Autoimmune: for instance, sarcoidosis.
-
Sporadic Creutzfeldt–Jakob disease
-
Toxic metabolic: for instance, Hashimoto's encepahitis, vitamin B1 deficiency, vitamin B12 deficiency, hyponatraemia and syndrome of inappropriate antidiuretic hormone secretion.
-
Vascular causes: cerebral vasculitis, cerebral venous thrombosis, small-vessel ischaemia
-
Late-onset white matter diseases: for instance, hereditary diffuse leukoencephalopathy with neuroaxonal spheroids.
-
Drugs: neuroleptic agents, benzodiazepines, recreational drugs.
-
Rapid progression of a chronic neurodegenerative disorder: Alzheimer's disease, frontotemporal dementia.
Among paraneoplastic causes, we should consider anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis. This characteristic syndrome develops in three stages. The first features a prodromal illness of fever, headache, nausea, gastrointestinal or respiratory symptoms. This is followed by psychiatric symptoms, short-term memory and speech problems. In the third phase, patients develop decreased responsiveness, abnormal movements (typically orolingual facial dyskinesias) and autonomic symptoms. MRI is abnormal in half of all cases, with T2 or FLAIR signal hyperintensity in the hippocampi, cerebellar or cerebral cortex, frontobasal and insular regions, basal ganglia, brainstem and rarely spinal cord. The CSF is abnormal in 80%; including a lymphocytic pleocytosis, and normal or with mildly increased protein. There are CSF-specific oligoclonal bands in 60%.13 This patient's age is against anti-NMDAR encephalitis, having a mean age of onset of 19 years. However, some cases are aged >70 years.13 Testing for NMDA (N-methyl-D-aspartate) receptor antibodies would help to exclude this diagnosis.
In the setting of a possible paraneoplastic disorder, one should consider other possible causes of confusion, such as hyponatraemia (from the syndrome of inappropriate antidiuretic hormone secretion) and hypercalcaemia. Hence, we would need to see his serum electrolytes, osmolality and serum calcium, corrected for serum albumin levels.
If we assume that the peripheral neuropathy is chronic and diabetic and unrelated to his presentation, what then could cause just the subacute dementia and ataxia? The differential diagnosis becomes much broader. However, we should first consider the possibility that the dementia and ataxia are, in fact, due to the hydrocephalus, as suggested on MRI.
Hydrocephalus
Ventricular enlargement follows from either a problem with CSF absorption (hydrocephalus) or from shrinkage of the brain tissue (cerebral atrophy) with neurodegenerative diseases and normal ageing. In practice, it is sometimes difficult to distinguish the reason for ventricular enlargement. Hydrocephalus is either communicating or non-communicating (ie, obstructive).
-
Obstructive hydrocephalus implies a blockage to CSF flow within the ventricular system. Without an obstructing mass, it is often caused by stenosis of the aqueduct linking the 3rd and 4th ventricles: hence, the 4th ventricle is not enlarged.
-
Communicating hydrocephalus implies that there is free passage of CSF through the ventricular system and out of the fourth ventricle, through the foramina of Luschka and Magendie. The problem is usually failure of CSF circulation or absorption in the subarachnoid space. In communicating hydrocephalus, the entire ventricular system, including the 4th ventricle is enlarged. Communicating hydrocephalus can occur acutely or chronically in patients who have, or have had, meningitis or subarachnoid haemorrhage.
A particular variety of chronic communicating hydrocephalus in the elderly has been called ‘normal pressure’, because the CSF pressure at a random single lumbar puncture is normal. It causes major diagnostic difficulties in elderly demented patients who are also off-balance and incontinent, as this particular triad of neurological problems, together with an enlarged ventricular system can be due to a neurodegenerative disease with cerebral atrophy, rather than to normal pressure hydrocephalus (NPH).14
Could this patient's problems be due to (communicating) hydrocephalus, as suggested by the brain MRI report, and could it be of the normal pressure variety? While his clinical syndrome (dementia+ataxia) is consistent with NPH, most patients with dementia and ataxia do not have NPH, even if they have the third component of the NPH triad (incontinence).14 Studies that simply document gait disturbance, dementia, incontinence and ventriculomegaly probably overestimate the prevalence of NPH, which is probably equal to that of progressive supranuclear palsy.14 Typically, NPH patients have subcortical/frontal cognitive dysfunction, most affected being memory, executive function, attention and psychomotor speed.15 The neuropsychological profile can improve after shunt insertion.16 However, other studies have found that while gait mght improve initially, incontinence and cognitive features do not improve following shunt insertion, suggesting a high degree of overdiagnosis.14 There is no definitive test for NPH. There are several supplemental tests to assist diagnosis, including the ‘CSF tap test’ (large-volume lumbar puncture), external CSF drainage by spinal drainage and CSF outflow determination with CSF infusion compliance studies.17 However, these tests have variable sensitivities and specificities. The CSF tap test has a low sensitivity 26–61%. Clinical response to CSF removal by spinal catheter has a higher sensitivity 50–100%, specificity 60–100% and positive predictive value 80–100%.18 However, it is an invasive procedure, requires expert management, and has a high complication rate.17
The rate of progression in this patient, over weeks, seems too rapid for NPH, which usually evolves over months to years. So if there really is communicating hydrocephalus, but is not of the normal pressure variety, what else could be causing it? One would have to think of infective or neoplastic causes.
Infective and neoplastic causes
Many CNS infections can cause cognitive decline. CSF results help to differentiate these, including CSF protein and white cell count and viral PCR (herpes simplex virus (HSV), cytomegalovirus (CMV), Epstein–Barr virus (EBV) and enterovirus). Against this in our patient, however, is the relatively subacute onset and lack of systemic features including fever. However, neurotropic viruses, such as enterovirus 71, can present with a subacute cognitive decline, which can be fatal.19 Also, measles can cause cognitive decline in elderly patients20 and mycoplasma can cause encephalitis.21 In immunocompromised patients, CMV and EBV infections can cause encephalitis22 and it would be important to know the patient's HIV status. There can also be a dementia-like presentation in Lyme disease.23
Chronic meningitis due to cryptococcal or tuberculous infection or leptomeningeal metastases may present with dementia and ataxia and can cause intracranial hypertension, either with (ie, hydrocephalus)24 ,25 or without ventricular enlargement (pseudotumour cerebri).26
Malignant meningitis presents with headache, dementia and ataxia in about half the cases.27 Meningeal lymphoma can cause raised intracranial pressure with papilloedema and/or hydrocephalus.28 Breast cancer (3%), small-cell lung cancer (6%), non-small-cell lung cancer (1%), gastrointestinal tumours (0.015–0.25%), carcinoma of unknown primary (3%) and melanoma (1.5%) can all produce neoplastic meningitis,27 and could cause raised intracranial pressure and hydrocephalus through decreased CSF absorption by the arachnoid villi. In this patient, the ‘pancreatic cysts’ identified on MRI could represent a possible neoplastic lesion. The rapid clinical decline is also consistent with this diagnosis.
On MRI, one would look for leptomeningeal enhancement. It would be important to know the lumbar CSF pressure, protein, glucose and differential white cell count, as well as cytology and flow cytometry to detect any malignant cells. CSF protein and white cell count may be the most useful tests in differentiating between viral, bacterial and cryptococcal meningitis, while CSF glucose concentration is less useful.29 A low glucose indicates a metabolically active process, but glucose can be low in both infective and neoplastic meningitis. It would also be important to know the results of cryptococcal antigen and tuberculosis (TB) PCR on CSF and cryptococcal serology on blood. The diagnosis of malignant meningitis might require several CSF examinations.
Could this be neurosyphilis, in one of its protean forms? The diagnosis depends on the results of serologic tests in blood and CSF; the interpretation of these results is often difficult—a positive result might not mean that syphilis is the cause. In the first instance, the blood syphilis serology should be checked and, if positive, the CSF examined. The diagnosis should be considered if the treponemal test is positive on CSF, there is an increased number of mononuclear cells, or if there is a positive venereal disease reference laboratory/rapid plasma reagin (VDRL/RPR) on the CSF. While a positive treponemal test on CSF does not confirm active neurosyphilis, a negative test excludes it.30
Neurological Whipple's disease is also possible. Patients present with cognitive decline, psychiatric symptoms, seizures and oculopalatal myoclonus. MRI should show one or more multinodular enhancing lesions. The CSF serology for Tropheryma whipplei is not always positive, and brain biopsy might be needed for diagnosis.31
One should also consider progressive multifocal leukoencephalopathy. Typically, patients present with weakness, sensory change, hemianopia, cognitive decline and imbalance and gait difficulties. MRI is critical to the diagnosis, usually showing areas of hyperintensity on T2 and FLAIR imaging and hypointensity on T1. Typically, the subcortical white matter, cerebellar peduncles, basal ganglia and thalamus are involved.32 The CSF should be checked for JC virus DNA.
Creutzfeldt-Jakob disease
Given the rapidity of the decline and cognitive and gait disturbance, it is important to consider sporadic Creutzfeldt–Jakob disease (sCJD). This can result in cognitive (39%), cerebellar (29%), behavioural (20%), constitutional (20%), sensory (11%), motor (9%) and visual (7%) symptoms.33 ,34 To fulfil WHO diagnostic criteria for CJD, patients require two of the following: myoclonus, pyramidal/extrapyramidal findings, visual or cerebellar deficits, and akinetic mutism. This patient had an extensor plantar on the right, fulfilling the pyramidal finding, and the gait problem could represent a cerebellar deficit. The EEG result would be important, specifically looking for periodic epileptiform discharges. Also the CSF looking for 14-3-3 protein and neurone-specific enolase. On CSF, one would also expect a mildly elevated protein, normal glucose and no leukocytosis.35 In sCJD, diffusion weighted (DWI) hyperintensities within the cortical and subcortical grey matter are highly sensitive and specific, and superior to either EEG, CSF neurone-specific enolase or 14-3-3 protein.36
Toxic/metabolic causes
We should also consider a toxic/metabolic cause. Hashimoto's encephalopathy can present with cognitive decline and ataxia, though more typically with a relapsing-remitting course and stroke-like episodes or with seizures.37 It would be important to know the thyroid function results and thyroid antibody results. Cognitive decline from intermittent exposure to carbon monoxide38 can present with slowing and memory decline, along with gait abnormalities39 and (unlike here) with globus pallidus abnormalities on MRI.40
Cerebral vasculitis
Isolated cerebral vasculitis can present with a subacute cognitive decline and non-specific symptoms. The diagnosis is difficult to make and relies on CSF examination, MRI/magnetic resonance angiography imaging and digital subtraction angiography. Both MRI and angiography can appear normal and have low diagnostic specificity. Brain biopsy is often required to help make the diagnosis.41
Cerebral venous thrombosis
Cerebral venous thrombosis of both the deep and superficial sinuses can present subacutely with non-specific signs and altered mentation and cognition. Cognitive decline can result from both white matter changes and venous hypertension, resulting in raised intracranial pressure.42 It would be important to review the patient's MRI and an MR venogram looking for thrombus, and to measure the CSF opening pressure.
Late-onset white matter diseases
Adult-onset leukodystrophies can present with subacute cognitive decline, gait problems and ataxia. In particular, metachromatic leukodystrophy, adrenoleukodystrophy, vanishing white matter disease and hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS) often present with mental deterioration and cognitive changes.43 ,44 However, the lack of white matter changes on MRI, spasticity or other neurological findings make leukodystrophy unlikely, although cognitive decline may precede the MRI changes in HDLS.44
Autoimmune disease
We should consider autoimmune causes, such as sarcoidosis or Sjögren's syndrome. Blood tests including an autoimmune and vasculitic screen and a chest x-ray for hilar lymphadenopathy would help. MRI can show white matter changes and meningeal involvement.
Drugs
Drugs can show, cause cognitive decline. In particular, neuroleptic agents can cause rapid cognitive decline in patients with dementia with Lewy bodies. However, this patient had no hallucinations, REM sleep behaviour disorder or extrapyramidal features. Despite this, it would be important to double check the drug history to ensure he received no neuroleptic agents.
Chronic neurodegenerative diseases
Chronic neurodegenerative diseases can present subacutely and be mistaken for a rapidly progressive dementia. In a retrospective study, 23% of patients diagnosed as rapidly progressive dementia had frontotemporal dementia—motor neurone disease, and 9% had Alzheimer's disease.45 In this patient, there was a 12-month history of decline, followed by 2 months of more rapid deterioration; it is possible that he had early Alzheimer's disease. In this situation, it would be important to perform a full septic screen (blood, chest x-ray and urine) to ensure one was not missing an infection that was worsening a chronic disorder. Careful examination of the MRI and volumetric MRI may be helpful in looking for focal atrophy that may point towards Alzheimer's disease or frontotemporal dementia.
Tests to be requested
To investigate this patient, based on the list of differential diagnoses, I would want the results of the following tests.
Blood: full blood count, electrolytes, liver function tests, thyroid function tests, thyroid antibodies, autoimmune and vasculitic screen, vitamin B12 and folate, homocysteine, methylmalonic acid, antineuronal antibodies, VGKC antibodies, NMDA receptor antibodies, syphilis serology and tumour markers (CEA, α fetoprotein, Ca19.9)
Imaging: repeat MRI brain with contrast, chest x-ray, whole body PET/CT scan.
CSF: pressure, white cell count, microscopy protein, glucose, oligoclonal bands, TB, Whipple's, HSV, CMV, EBV and enterovirus PCR, cryptococcal antigen, cytology, flow cytometry and VDRL.
EEG.
Results of tests
Blood
-
Electrolytes (sodium 136 mmol/l, potassium 4.5 mmol/l, chloride 98 mmol/l urea 7.6 mmol/l, creatinine 86 umol/l), full blood count and liver function tests: normal.
-
Autoimmune and vasculitic screen (including ANA, ENA, double-stranded DNA, ANCA, rheumatoid factor, immunoglobulins, immune electrophoresis and cryoglobulins): negative.
-
Vitamin B12 (285 pmol/l), serum folate (20.2 nmol/l), thyroid function (TSH 3.520 mIU/l, FT4 14.4pmol/l): normal; thyroid antibodies negative.
-
Homocysteine, methylmalonic acid: not done.
-
Hepatitis B, C and HIV serology: negative.
-
Syphilis antibodies EIA screen: non-reactive.
-
Tumour markers CEA: 9.5 ug/l, Ca19.9: 85 Ku/l, Ca15.3: 17 Ku/l, α-fetoprotein: 1.3 IU/ml): all negative.
-
Antineuronal antibodies to Yo, Hu and Ri: negative. VGKC and NMDA receptor antibodies were not done.
CSF
-
Pressure 170 mm H2O, protein 1.31 g/l (n= <0.40), glucose 2.0 mmol/l (serum glucose 7 mmol/l), four white cells, 11 red cells.
-
Oligoclonal bands: negative in both CSF and serum.
-
CSF cytology: mild increase in mixed inflammatory cells.
-
Flow cytometry: very few viable CD45+ cells, remaining cells are predominantly CD3+CD4+.
-
CSF 14-3-3 protein: atypical positive result, but not typical for sCJD, given raised CSF protein (>1.0 g/l).
-
Neurone-specific enolase positive.
-
TB, HSV, EBV, enterovirus PCR negative.
-
Cryptococcal antigen negative.
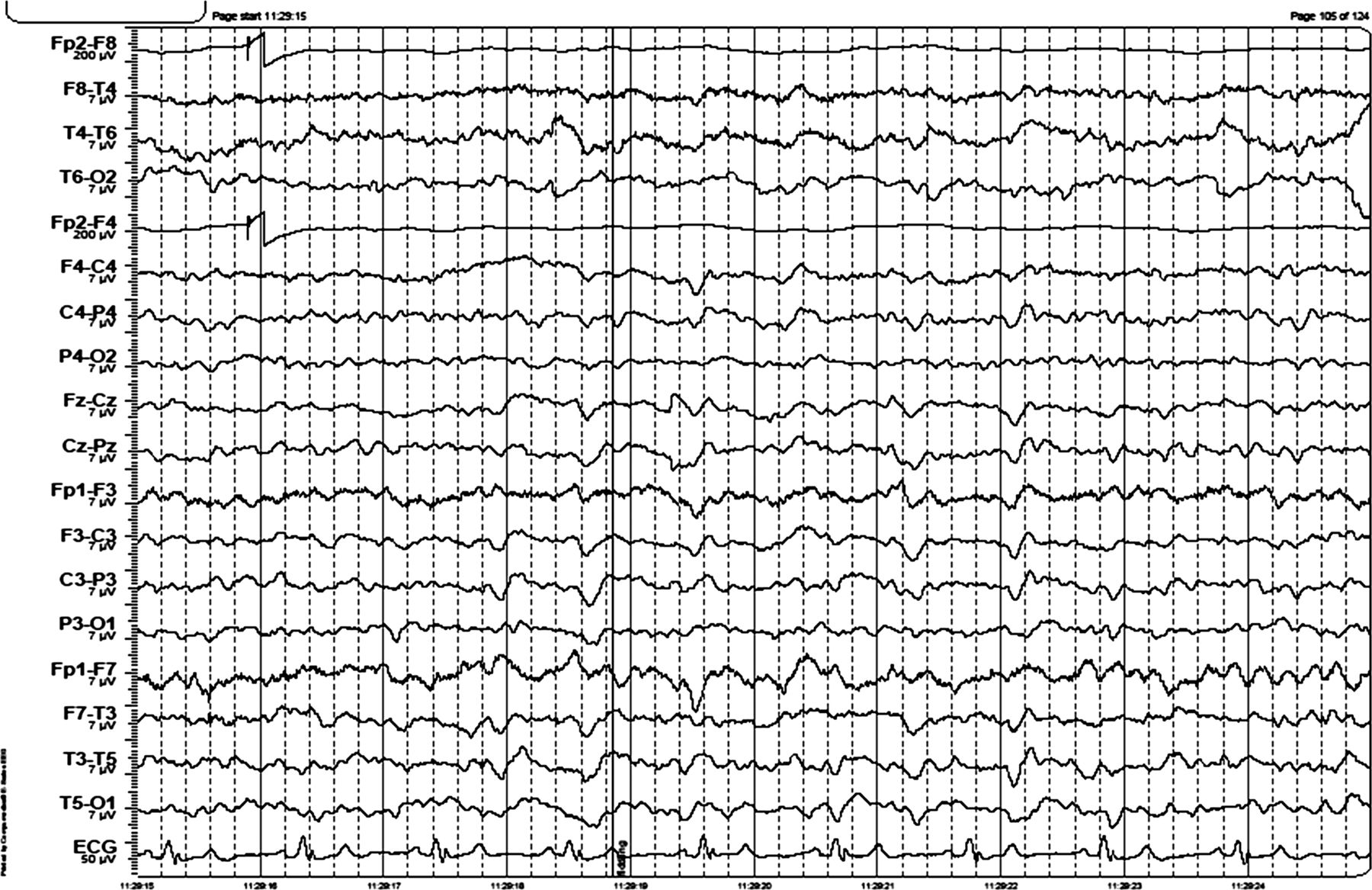
EEG: see figure 2.
Chest x-ray showed a suspicious lesion at the right lung base. Brain MRI was not repeated.
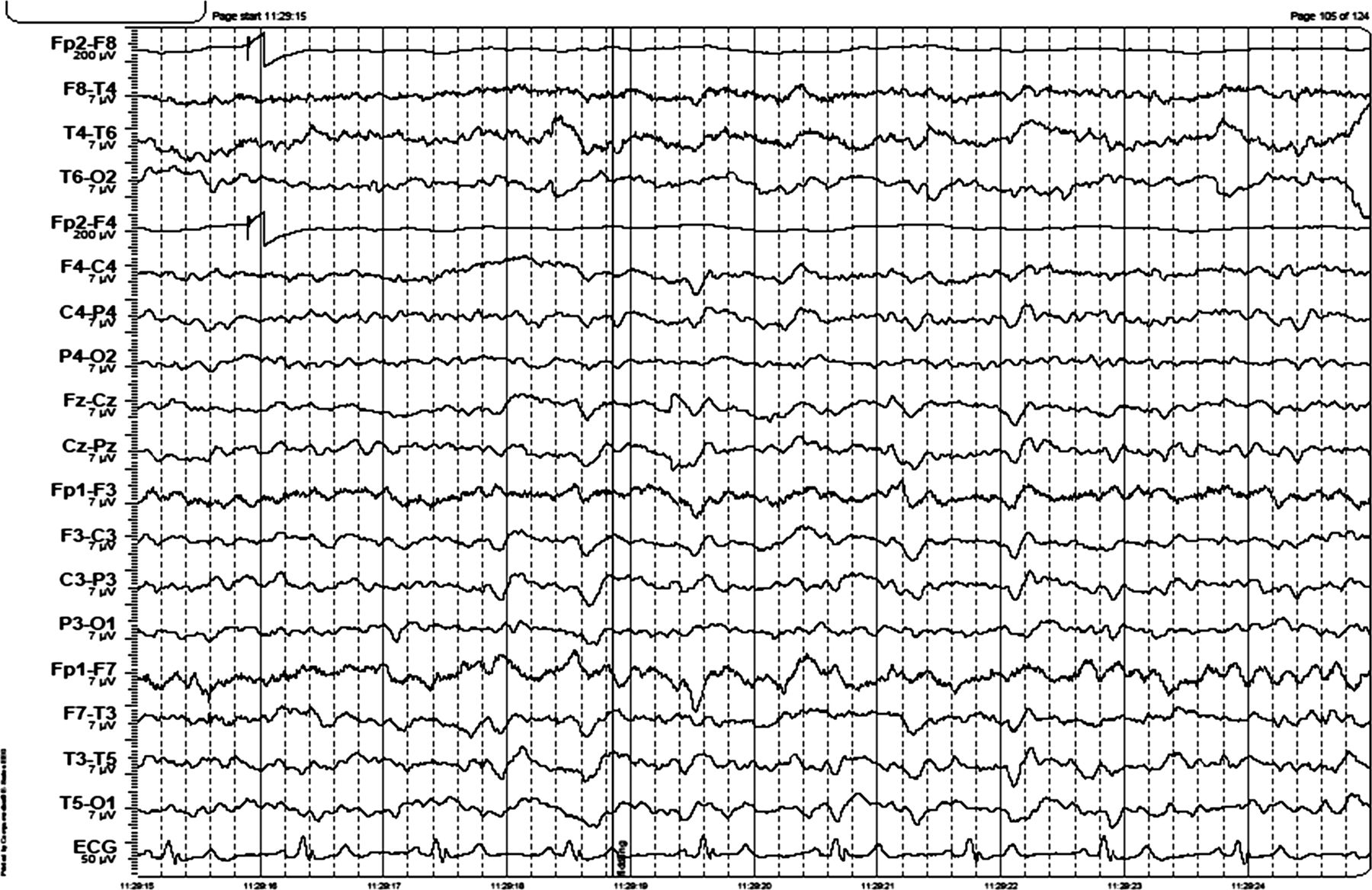
EEG shows intermittent generalised 3–4 Hz activity lasting 1 s and continuous generalised 5 Hz activity. The EEG was reactive to eye opening. There are left hemisphere quasiperiodic triphasic waves.
Discussion Part B (Dr GM Halmagyi)
While the EEG and the CSF 14-3-3 protein result give soft support for a diagnosis of CJD, the high CSF protein and the low glucose46 are against the diagnosis and also against the diagnosis of paraneoplastic limbic encephalitis: they more support a meningitic process—even with a normal white cell count. The negative CSF TB-PCR and cryptococcal antigen make it unlikely that the ‘suspicious’ lung lesion is causing tuberculous or cryptococcal meningitis, and I suspect a primary or secondary cancer. I would want a whole body PET/CT and then a biopsy of the lung lesion.
Progress
CT chest, abdomen and pelvis showed an infiltrating lesion at the right lung base, suggesting a cancer. There were masses in both adrenal glands suggesting metastases. PET scan was not done.
The patient continued to decline over the next 2 weeks, with decreased conscious level and was unable to walk. He was admitted to hospital and underwent bronchoscopy which showed extrinsic narrowing of the bronchus intermedius, the right middle lobe bronchus and the superior segmental bronchus of the lower lobe. Bronchial brushings showed malignant cells consistent with large-cell carcinoma of the lung. The patient and his family declined further treatment. Over the following weeks he became less responsive, the ventricles continued to enlarge and periventricular CSF transudation increased (figure 3). He died 3 weeks following his initial presentation to the neurologist.
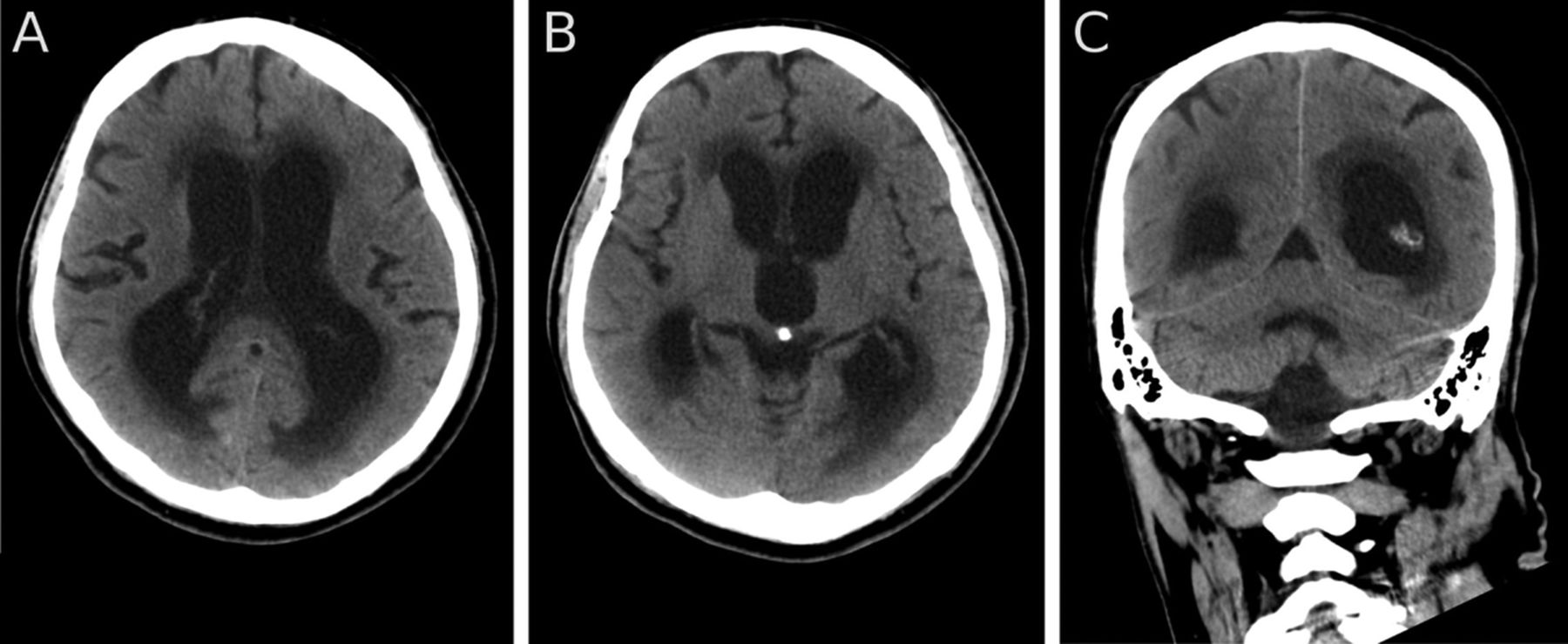
Axial (A, B) and coronal (C) CT 2 weeks after the MRI (figure 1) and 1 week before death showing an increase in the size of the ventricles and in CSF exudation.
Dr Halmagyi's diagnosis: malignant meningitis.
Pathology (Dr M Rodriguez)
A full autopsy was performed. There was a 4 cm diameter moderately well-differentiated adenocarcinoma in the apex of the lower lobe of the right lung. The right hilar lymph nodes were enlarged, matted together and contained metastatic carcinoma. There were no adrenal masses.
The brain weighed 1190 g. There was >90% stenosis of the distal vertebral artery by atheroma and a retrocerebellar arachnoid cyst. The foramina of Magendie and Luschka were patent. There was thickening and yellow/orange discolouration of the basal leptomeninges. There was hydrocephalus with symmetric dilatation of the lateral and third ventricles (figure 4) as well as enlargement of the aqueduct and fourth ventricle. The septum pellucidum was neither fenestrated nor thinned. The periventricular white matter was pale and soft.

Coronal section of cerebrum at the level of the lateral geniculate nuclei showing marked symmetric enlargement of the lateral and third ventricles. The white matter pallor adjacent to the ventricles is due to periventricular oedema secondary to transependymal spread of cerebrospinal fluid.
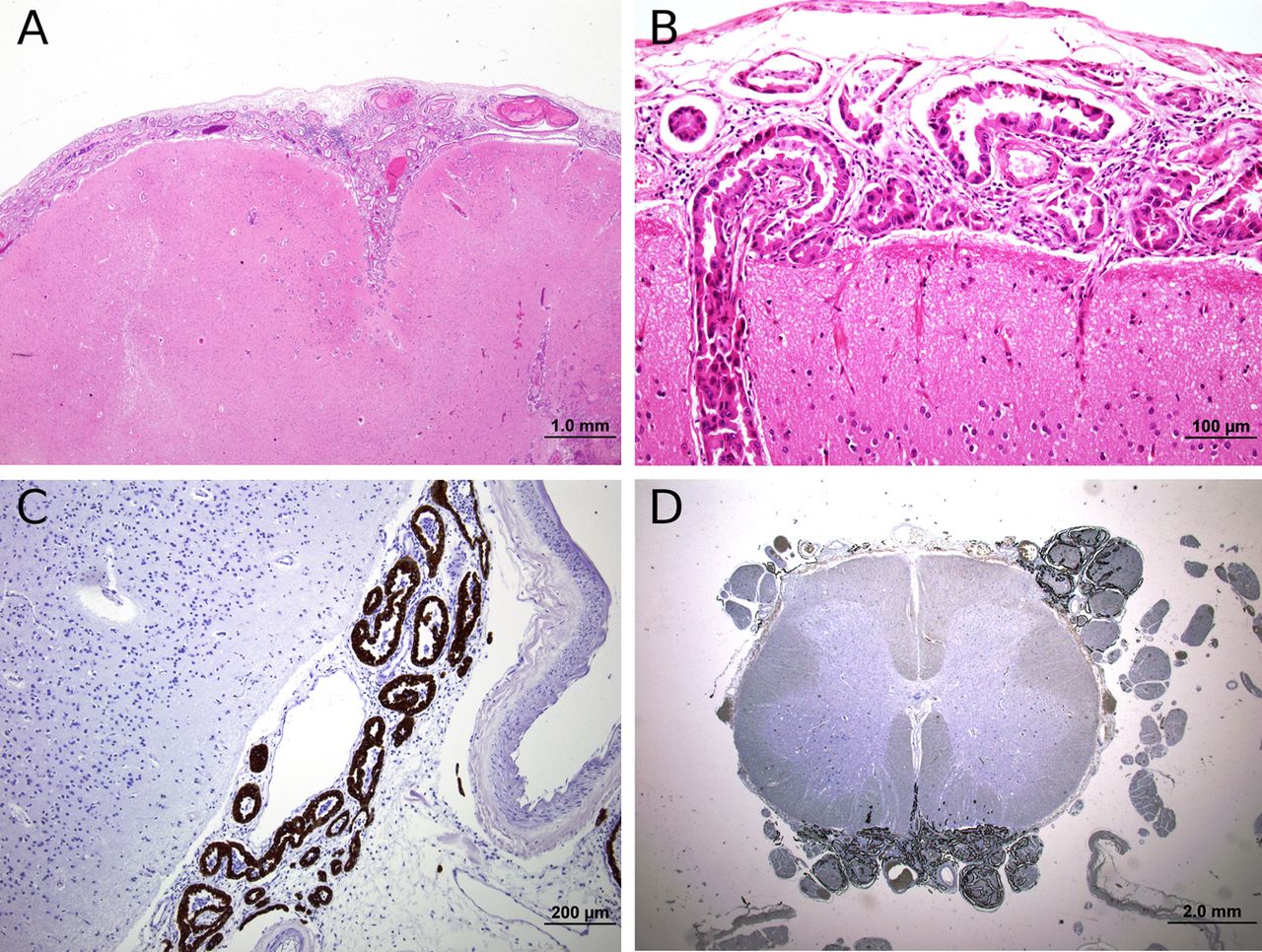
Microscopically, there was widespread leptomeningeal carcinomatosis in almost every section examined, with extension into the Virchow–Robin spaces, and there were several microscopic foci of superficial parenchymal invasion. The tumour resembled the pulmonary adenocarcinoma (figure 5A,B). Tumour also infiltrated the extra-axial segments of several cranial nerves and the dorsal and ventral roots in the spinal cord (figure 5C,D), most marked in the lumbar region where several neurones in the ventral horns show central chromatolysis. There were also multiple areas of recent infarction, predominantly in the cerebral cortex, often associated with tumour infiltration into the Virchow–Robin spaces.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Moderately well differentiated adenocarcinoma in subarachnoid space (H&E). Original magnification ×100 (B) Adenocarcinoma in subarachnoid space extending into Virchow- Robins space (H&E). Original magnification ×400 (C) Lumbar spinal cord with leptomeningeal carcinomatosis with dorsal and ventral root infiltration. ×12.5 (cytokeratin 8 immunoperoxidase). Original magnification ×12.5. (D) Lumbar ventral roots showing tumour infiltration (cytokeratin 8 immunoperoxidase). Original magnification ×400.
The mild small-vessel disease in the cerebral white matter was not severe enough to explain either the MRI or the clinical features. There was widespread microglial activation, but no inflammation to support a diagnosis of vasculitis or a paraneoplastic or autoimmune encephalitis. There were also no features of infection, prion disease, acute or chronic Wernicke's encephalopathy, subacute combined degeneration or chronic neurodegenerative disease including frontotemporal lobar degeneration, Alzheimer's disease or dementia with Lewy body disease. The hydrocephalus was most likely due to obstruction of CSF flow in the subarachnoid space secondary to leptomenigeal carcinomatosis from the right lung adenocarcinoma.
Concluding remarks
This patient presented with dementia, ataxia and communicating hydrocephalus due to malignant meningitis from a primary lung adenocarcinoma. Neoplastic meningitis only rarely results from a non-small-cell lung cancer, and occurrs in only 1% of patients.27 In a series of 640 patients with malignant meningitis, only 37 had hydrocephalus.47
The key to the diagnosis of neoplastic meningitis is contrast-enhanced MRI of both brain and spine, and CSF cytology. In a study of 137 patients with clinical symptoms of neoplastic meningitis, imaging was abnormal in 55%, including leptomeningeal, subependymal, dural or cranial nerve enhancement; superficial cerebral lesions or communicating hydrocephalus.48 However, contrast-enhanced brain MRI might not be positive. A recent study of 44 patients with neoplastic meningitis found a sensitivity of 45% for detecting meningeal involvement: the sensitivity was higher for solid tumours (84.6%) and lower for leukaemia (20%) and lymphoma (37.5%).49 In this case, neither MRI spine or contrast-enhanced brain MRI were performed, but the fact that the brain and spine were macroscopically normal at autopsy suggests there would not have been unequivocal contrast enhancement. It is possible that the patient did develop focal signs suggesting spinal involvement towards the end of his illness, but by then he was too obtunded to examine clinically. However, there are case reports of malignancy, particularly lymphoma, with leptomeningeal involvement of spinal nerve roots that have no localising signs,28 and in some cases normal spinal imaging.50
The sensitivity of CSF cytology in malignant meningitis is influenced by sample volume and time to fixation.46 In this patient, the clues to a meningeal process were a high protein and low glucose, but the cytology showed no malignant cells. The optimum CSF cytology sampling technique is to take at least 10 ml of CSF and to collect the sample into a tube with formalin fixative before sending it to the laboratory.51 Cytology has a sensitivity of 93% for detecting neoplastic meningitis.49 In this patient, the diagnosis would have been made sooner had a chest x-ray been requested at his initial presentation.
Acknowledgments
Our thanks to Dr GD Parker for interpreting the radiology, and to Dr Brian Elston for carrying out the autopsy.
References
Footnotes
-
A clinicopathological conference held at the Royal Prince Alfred Hospital, Sydney in March 2011
-
Contributors RMA: case presentation and manuscript preparation. Takes overall responsibility for content of manuscript. GMH: discussant of manuscript, manuscript preparation. MLR: pathology presentation.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed. Reviewed by Simon Lewis, Sydney, Australia.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Other content recommended for you
- Community-acquired bacterial meningitis in adults
- An exotic cause for confusion in the garden
- Idiopathic normal pressure hydrocephalus: historical context and a contemporary guide
- Meningeal derived cerebrospinal fluid proteins in different forms of dementia: is a meningopathy involved in normal pressure hydrocephalus?
- Is normal pressure hydrocephalus a valid concept in 2002? A reappraisal in five questions and proposal for a new designation of the syndrome as “chronic hydrocephalus”
- Hydrocephalus due to extreme dilation of Virchow-Robin spaces
- Isolated leptomeningeal carcinomatosis and possible fungal meningitis as late sequelae of oesophageal adenocarcinoma
- Comparison between the lumbar infusion and CSF tap tests to predict outcome after shunt surgery in suspected normal pressure hydrocephalus
- Organic neuropsychiatry: a treatable cause of suicidal behaviour
- Chronic meningitis, seizures and myoclonus